Adrenalectomy—Open and Minimally Invasive
L. Michael Brunt
Arthur Rawlings
Tumors of the adrenal gland may present in a variety of clinical manifestations depending on the underlying pathophysiology. Surgical resection is the preferred treatment for a primary adrenal mass that is hormonally functional or malignant. Accurate diagnosis and localization through a systematic approach with biochemical testing and imaging techniques are essential in selecting patients for operation. Several open and laparoscopic approaches are available to remove the adrenal gland, each with its own advantages in terms of exposure, degree of invasiveness, ability to explore the entire abdomen, and patient outcomes. Tumor size, functionality, bilaterality or extra-adrenal location, malignant potential, individual patient characteristics, and surgeon experience all play an important role in determining the type of procedure utilized. Patients with hormonally functional tumors should undergo adequate preoperative preparation to minimize intraoperative complications. Proper selection of patients for operation, a thorough knowledge of adrenal anatomy, and a meticulous and hemostatic extra-adrenal dissection technique are imperative to optimize outcomes, especially in the minimally invasive setting.
The adrenal glands are retroperitoneal organs immediately superior to the kidneys. These glands are slightly nodular with a firm texture and are surrounded by a layer of areolar connective tissue. Each gland weighs approximately 4 to 5 g in the adult and has a golden yellow-orange color distinct from the pale yellow retroperitoneal fat. The left adrenal gland is bordered inferiorly by the left kidney and left renal vein, superiorly and posteriorly by the diaphragm, anteriorly by the tail of the pancreas, and medially by the spleen and aorta. The right adrenal gland is bordered inferiorly by the right kidney; superiorly, posteriorly, and laterally by the diaphragm; anteriorly by the liver; and medially by the inferior vena cava (IVC; Fig. 1).
The arterial blood supply to the adrenal gland is derived mainly from the inferior phrenic and renal arteries as well as directly off the aorta. Occasionally, intercostal and ovarian vessels may contribute as well. Rather than one or two main arteries, the distribution is primarily one of multiple small branches that enter the superior, medial, and inferior aspects of the gland (Fig. 2). Venous drainage of the left adrenal is via the left adrenal vein, which arises from the inferomedial aspect of the gland and empties into the left renal vein. The inferior
phrenic vein usually joins the left adrenal vein above its entry into the renal vein. Blood from the right adrenal gland empties directly into the IVC via the short central adrenal vein that arises from the medial aspect of the gland. Accessory adrenal veins entering the vena cava or hepatic veins may be present on the right side. Lymphatic drainage from the adrenal gland is into adjacent pericaval and periaortic lymph nodes, which is important during resection of a malignant tumor.
phrenic vein usually joins the left adrenal vein above its entry into the renal vein. Blood from the right adrenal gland empties directly into the IVC via the short central adrenal vein that arises from the medial aspect of the gland. Accessory adrenal veins entering the vena cava or hepatic veins may be present on the right side. Lymphatic drainage from the adrenal gland is into adjacent pericaval and periaortic lymph nodes, which is important during resection of a malignant tumor.
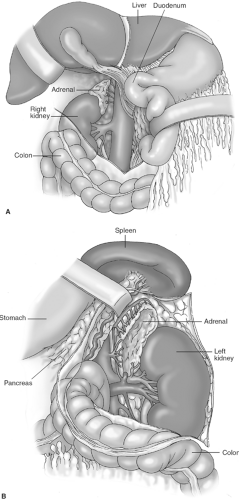 Fig. 1. Anatomic relationships of the adrenal glands to surrounding structures. |
The adrenal gland is divided into two distinct components: the cortex and the medulla. The adrenal cortex is the site of production of mineralocorticoids (aldosterone), glucocorticoids (cortisone), and sex steroids. The medulla contains chromaffin cells, which secrete the catecholamines epinephrine and norepinephrine and also dopamine.
Adrenal tumors may come to attention because of clinical signs and symptoms of hormone hypersecretion, because of local symptoms of pain due to a large mass, or as a lesion discovered incidentally during cross-sectional imaging done for other reasons. Appropriate diagnosis and localization are imperative for successful operative planning and treatment. The various indications for adrenalectomy are given in Table 1. Algorithms for biochemical testing as well as radiographic imaging are available for differentiating the various causes of adrenal lesions including aldosteronoma, Cushing syndrome, pheochromocytoma, adrenal cortical carcinoma, metastatic disease of the adrenal glands, and adrenal incidentaloma. A summary of the biochemical evaluation and preoperative preparation used for the treatment of these adrenal lesions is described in Table 2.
Aldosteronoma
Primary hyperaldosteronism is the most common cause of secondary hypertension and has a much higher prevalence rate in the population than previously appreciated, occurring in 8% to 12% of hypertensive patients. Although the classic findings in primary hyperaldosteronism are hypertension and hypokalemia, many patients with this diagnosis have a normal serum potassium level. Therefore, any patient who has hypertension with an early age of onset that is difficult to control or refractory to medical management should be considered for this diagnosis, regardless of the serum potassium level. Because endogenous hyperaldosteronism suppresses renin secretion, biochemical screening entails simultaneous measurement of both plasma aldosterone and renin levels. A plasma aldosterone concentration (PAC)–to–plasma renin activity (PRA) ratio greater than 20:25 in the setting of an absolute PAC greater than 15 ng/dL is consistent with this diagnosis and should be evaluated further. Confirmatory testing consists of demonstrating elevated
24-hour urine aldosterone levels (>12 μg/24 h) while on a high-sodium diet or after intravenous saline loading. Other biochemical findings include an elevated urinary potassium excretion rate (>30 mEq/24 h).
24-hour urine aldosterone levels (>12 μg/24 h) while on a high-sodium diet or after intravenous saline loading. Other biochemical findings include an elevated urinary potassium excretion rate (>30 mEq/24 h).
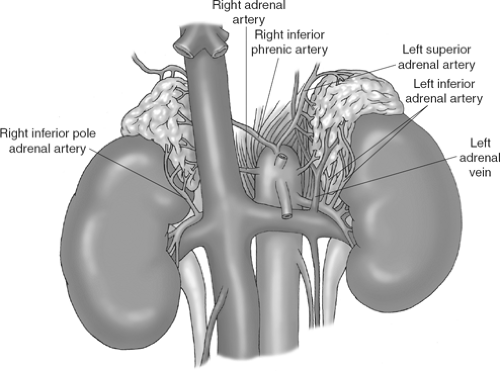 Fig. 2. Blood supply to the adrenals. |
Table 1 Indications for Adrenalectomy | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The most common causes of primary hyperaldosteronism are aldosterone-producing adenoma (65%) and idiopathic cortical adrenal hyperplasia (35%). Aldosterone-secreting adrenal carcinomas are rare. Differentiation of these causes is critical in directing therapy since the preferred treatment of an aldosteronoma is adrenalectomy, whereas idiopathic hyperaldosteronism from cortical hyperplasia is treated medically with the aldosterone antagonist spironolactone. Adrenal cross-sectional imaging with thin-cut (3-mm image slices) computed tomography (CT) is indicated once primary aldosteronism has been confirmed biochemically. Younger patients (under age 40 to 50) with a discrete unilateral macroadenoma (>1 cm) and a normal contralateral adrenal may undergo adrenalectomy without further testing. All other patients should undergo adrenal vein sampling for cortisol and aldosterone to determine if there is a lateralizing source of increased aldosterone production.
Most aldosteronomas are small, less than or equal to 1 to 2 cm in size, with a golden orange color. These tumors are rarely malignant and are ideally suited for laparoscopic excision. Spironolactone may be given preoperatively to control hypertension but should not be administered until the biochemical evaluation, including adrenal vein sampling, is complete. Patients should also have potassium levels repleted and, in long-standing cases, assessment of cardiac function and renal insufficiency prior to surgery.
Table 2 Diagnosis and Preoperative Preparation of Common Adrenal Tumors | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
Cushing Syndrome
Cushing syndrome results from excessive cortisol secretion from a variety of pathologic processes that is not controlled by normal regulatory mechanisms. Overproduction of glucocorticoids may lead to development of characteristic features such as truncal obesity, moon facies, plethora,
hirsutism, abdominal striae, acne, and the presence of a “buffalo hump.” Hypertension and diabetes are also commonly present.
hirsutism, abdominal striae, acne, and the presence of a “buffalo hump.” Hypertension and diabetes are also commonly present.
Causes of Cushing syndrome can be divided into those that are adrenocorticotropic hormone (ACTH) dependent and those that are ACTH independent. Most ACTH-dependent cases are related to Cushing disease (due to excessive pituitary production of ACTH), while other cases may result from ectopic production of ACTH by other tumors. ACTH-independent causes include primary adrenocortical diseases such as cortisol-producing adenoma, adrenocortical carcinoma, and adrenal hyperplasia. Suspected Cushing syndrome should be evaluated with measurement of 24-hour urine-free cortisol levels. A single-dose dexamethasone test can also be used to screen for Cushing syndrome. In this test, 1 mg of dexamethasone is given at 11:00 PM, and a morning (8:00 AM) cortisol level is obtained. Normal individuals are able to suppress plasma cortisol to less than 3 μg/dL, whereas patients with Cushing syndrome fail to suppress. Late-night (11:00 PM) salivary cortisol levels are also highly sensitive and specific in screening for Cushing syndrome and are increasingly being used by some groups.
Once Cushing syndrome is confirmed biochemically, plasma ACTH levels should be measured to differentiate ACTH-dependent from ACTH-independent causes. A low plasma ACTH suggests adrenocortical disease and should be evaluated further with CT. Patients with normal or elevated plasma ACTH levels should have pituitary imaging (pituitary magnetic resonance imaging (MRI), inferior petrosal sinus sampling) to evaluate for Cushing disease as the cause and to localize the tumor. Adrenalectomy is the treatment for Cushing syndrome from an adrenocortical tumor. Patients with Cushing disease who fail treatment of the pituitary lesion may benefit from bilateral adrenalectomy. It is important to administer stress doses of steroids to these patients in the preoperative period since they may not be physiologically capable of responding to stress with endogenous glucocorticoids. Patients undergoing bilateral adrenalectomy should also be given replacement mineralocorticoids postoperatively.
Subclinical Cushing Syndrome
Subclinical Cushing syndrome (SCS) is a condition is which there is autonomous secretion of cortisol but without the typical clinical signs of classic Cushing syndrome. SCS is most commonly identified in the setting of an adrenal incidentaloma. Laboratory findings include lack of suppressibility of cortisol secretion with dexamethasone, loss of the diurnal variation in cortisol secretion, low or suppressed plasma ACTH levels, and a blunted response of ACTH to corticotrophin-releasing hormone. Elevation of 24-hour urine-free cortisol levels is seen in up to 50% of patients with SCS but the degree of elevation is usually mild. Although patients with subclinical Cushing have a higher incidence of hypertension, obesity, and diabetes than controls, the natural history is not well characterized. In one study, progression to overt Cushing syndrome at 1-year follow-up was 12.5%. Adrenalectomy is indicated for patients with this condition who are an acceptable risk for surgery. SCS is important to recognize in the setting of adrenal incidentaloma as these patients may develop adrenal insufficiency after adrenalectomy if glucocorticoid replacement is not administered.
Pheochromocytoma
Pheochromocytomas are rare tumors that arise from the chromaffin cells of the adrenal medulla. Approximately 10% of tumors in adults (up to 35% in children) arise in extra-adrenal locations such as the organ of Zuckerkandl, bladder, renal hilum, or rarely elsewhere along the sympathetic chain. While most pheochromocytomas are unilateral and benign, approximately 10% are bilateral in location (seen more commonly in hereditary endocrine syndromes) and up to 10% may be malignant. These tumors are generally functional and secrete excessive catecholamines including epinephrine, norepinephrine, and dopamine. Patients often present with symptoms of episodic spells consisting of headaches, diaphoresis, and palpitations in association with marked hypertension.
Screening for pheochromocytoma consists of measurement of either plasma fractionated metanephrines or 24-hour urinary metanephrines and catecholamines (epinephrine, norepinephrine, and dopamine). Indications for screening for pheochromocytoma include refractory or accelerated hypertension, labile hypertension, hypertensive paroxysms during anesthesia or sedation, adrenal incidentaloma, paradoxic hypertension in response to beta-blockers, and familial screening for hereditary endocrinopathies. Patients with biochemical evidence suggestive of pheochromocytoma should undergo cross-sectional imaging. T2-weighted MRI sequences often show a bright appearance of the tumor that is characteristic for pheochromocytoma (adrenal mass/liver image intensity ratio >3.0). 123I-metaiodobenzylguanidine (MIBG) scanning may occasionally be useful in localizing functional, extra-adrenal, or metastatic tumors. 123I-MIBG is not warranted in patients with uncomplicated pheochromocytomas that are localized on CT or MRI, as it is expensive and rarely alters treatment in this setting.
Once the diagnosis of a pheochromocytoma is made, the patient should be placed on alpha-blockade with phenoxybenzamine to control hypertension and dilate the intravascular space, and should be instructed to drink ample fluids. Beta-blockade may be added if the patient develops tachycardia on phenoxybenzamine or has a predominately epinephrine-secreting tumor. For further details, see the section on Patient Preparation below.
Adrenocortical Carcinoma
Adrenocortical carcinoma is a rare malignancy with an annual incidence of less than two cases per million individuals. It carries a poor prognosis, as many patients (up to 40%) have advanced or metastatic disease at the time of presentation. Adrenocortical cancers are usually large tumors with an average diameter of around 12 cm and may be functional or nonfunctional. Malignancy should be suspected in any adrenal cortical tumor greater than 6 cm in diameter, as the incidence of malignancy increases with increasing tumor size. Nonfunctional tumors may present as abdominal or back pain, weight loss, malaise, or hematuria. A majority of adrenal cancers (approximately 60%) are functional, however, with symptoms of Cushing syndrome, virilization, or both.
Preoperative considerations and preparation are similar to those for other functioning adrenal tumors. CT scanning or MRI is necessary to fully evaluate the extent of disease as well as possible involvement of major vascular structures and regional or distant metastases. Surgical resection remains the only potentially curative treatment.
Adrenal Incidentaloma
The most common adrenal mass encountered by the clinician is the adrenal incidentaloma discovered during abdominal imaging for a non–adrenal-related workup. The incidence of finding an adrenal mass on abdominal CT scanning ranges from 0.4% to 4.4%. The key factors in evaluating an adrenal incidentaloma are to characterize its size, functionality, and risk of malignancy. Functional masses should be removed regardless of size. Nonfunctional tumors 4 to 5 cm or larger should be removed, as should lesions in which the imaging characteristics are atypical for an adenoma. The biochemical evaluation of the adrenal incidentaloma should include measurement of plasma
fractionated metanephrines or 24-hour urine metanephrines and catecholamines to exclude a pheochromocytoma and a single low-dose (1-mg) dexamethasone test to evaluate for subclinical hypercortisolism. Plasma aldosterone and renin levels should be done only if the patient is hypertensive or hypokalemic.
fractionated metanephrines or 24-hour urine metanephrines and catecholamines to exclude a pheochromocytoma and a single low-dose (1-mg) dexamethasone test to evaluate for subclinical hypercortisolism. Plasma aldosterone and renin levels should be done only if the patient is hypertensive or hypokalemic.
The most common adrenal lesion discovered as an incidentaloma is a nonfunctioning cortical adenoma. Adrenal myelolipomas are benign lesions composed of fat and bone marrow elements that can be diagnosed by their typical radiographic appearance. They do not need to be removed unless they are enlarging or become symptomatic, such as from hemorrhage into the lesion. Nonfunctioning tumors less than 4 cm should be followed with serial imaging at 4 and 12 months.
Occasionally other primary tumors such as lung cancer, breast cancer, renal cell carcinoma, and melanomas metastasize to the adrenal glands. In addition to CT or MRI, positron emission tomography (PET) should be done in potentially resectable cases to exclude metastatic disease in other sites. Surgical resection may be indicated for selected patients with an isolated adrenal metastasis. Percutaneous biopsy of suspected adrenal metastases should be reserved for patients who are not candidates for surgical resection and in whom the results of biopsy will impact therapy.
Patient Preparation
Prior to adrenalectomy, patients should have electrolyte abnormalities such as hypokalemia corrected. Hypertension should be controlled medically and patients with pheochromocytomas should receive 7 to 10 days of preoperative alpha-receptor blockade with phenoxybenzamine to avoid hypertensive exacerbations intraoperatively. Phenoxybenzamine is typically started in a dose of 10 mg twice daily and the dose is increased by 10 to 20 mg/d every 2 to 3 days until the blood pressure is well controlled and the patient is mildly orthostatic. Some patients with symptomatic pheochromocytomas in hypertensive crisis will need to be blocked while in the hospital, but for many patients the blockade can be managed on an outpatient basis. During this time, patients should be instructed to drink ample fluids to allow volume expansion as the alpha-blockade proceeds. Our practice has been to admit patients to the hospital the day before the scheduled adrenalectomy to increase the blockade and allow monitoring of orthostatic vital signs and urinary output. A total dose of phenoxybenzamine of 40 to 60 mg/d is sufficient to block most patients. Intraoperatively, patients with vasoactive pheochromocytomas should have continuous blood pressure monitoring via an arterial line. Intraoperative fluid resuscitation is crucial to avoid hypotension after the tumor has been removed.
Patients with Cushing syndrome should receive intravenous steroids perioperatively and will require maintenance doses of steroids for 6 months or longer postoperatively. Mechanical bowel cleansing is not routinely given prior to adrenalectomy.
Selection of Operative Approach
The retroperitoneal location of the adrenals makes them accessible via a variety of surgical approaches: transabdominal, retroperitoneal, or through the flank, and using either laparoscopic or open techniques. The choice of surgical approach in a given patient depends on the size and functionality of the lesion, the degree of exposure needed, the likelihood that the tumor is malignant, and the surgeon’s experience (Table 3). For most patients with an adrenal tumor, laparoscopic adrenalectomy is appropriate and has become preferred over the various open approaches. This approach has been associated with less pain, a shorter hospitalization, faster return to unrestricted activities, and less morbidity than open adrenalectomy. Late complications such as incisional hernia and chronic incisional pain are also less common with the laparoscopic approach.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


