Adrenal Gland Disorders
KEY CONCEPTS
![]() Glucocorticoid secretion from the adrenal cortex is stimulated by adrenocorticotropic hormone (ACTH) or corticotropin that is released from the anterior pituitary in response to the hypothalamic-mediated release of corticotropin-releasing hormone (CRH).
Glucocorticoid secretion from the adrenal cortex is stimulated by adrenocorticotropic hormone (ACTH) or corticotropin that is released from the anterior pituitary in response to the hypothalamic-mediated release of corticotropin-releasing hormone (CRH).
![]() To ensure the proper treatment of Cushing’s syndrome, diagnostic procedures should (a) establish the presence of hypercortisolism and (b) discover the underlying etiology of the disease.
To ensure the proper treatment of Cushing’s syndrome, diagnostic procedures should (a) establish the presence of hypercortisolism and (b) discover the underlying etiology of the disease.
![]() The rationale for treating Cushing’s syndrome is to reduce the morbidity and mortality resulting from disorders such as diabetes mellitus, cardiovascular disease, and electrolyte abnormalities.
The rationale for treating Cushing’s syndrome is to reduce the morbidity and mortality resulting from disorders such as diabetes mellitus, cardiovascular disease, and electrolyte abnormalities.
![]() The treatment of choice for both ACTH-dependent and ACTH-independent Cushing’s syndrome is surgery, whereas pharmacologic agents are reserved for adjunctive therapy, refractory cases, or inoperable disease.
The treatment of choice for both ACTH-dependent and ACTH-independent Cushing’s syndrome is surgery, whereas pharmacologic agents are reserved for adjunctive therapy, refractory cases, or inoperable disease.
![]() Pharmacologic agents that may be used to manage the patient with Cushing’s syndrome include steroidogenesis inhibitors, adrenolytic agents, neuromodulators of ACTH release, and glucocorticoid-receptor blocking agents.
Pharmacologic agents that may be used to manage the patient with Cushing’s syndrome include steroidogenesis inhibitors, adrenolytic agents, neuromodulators of ACTH release, and glucocorticoid-receptor blocking agents.
![]() Spironolactone, a competitive aldosterone receptor antagonist, is the drug of choice in bilateral adrenal hyperplasia (BAH)–dependent hyperaldosteronism.
Spironolactone, a competitive aldosterone receptor antagonist, is the drug of choice in bilateral adrenal hyperplasia (BAH)–dependent hyperaldosteronism.
![]() Addison’s disease (primary adrenal insufficiency) is a deficiency in cortisol, aldosterone, and various androgens resulting from the loss of function of all regions of the adrenal cortex.
Addison’s disease (primary adrenal insufficiency) is a deficiency in cortisol, aldosterone, and various androgens resulting from the loss of function of all regions of the adrenal cortex.
![]() Secondary adrenal insufficiency usually results from exogenous steroid use, leading to hypothalamic–pituitary–adrenal (HPA)–axis suppression followed by a decrease in ACTH release, and low levels of androgens and cortisol.
Secondary adrenal insufficiency usually results from exogenous steroid use, leading to hypothalamic–pituitary–adrenal (HPA)–axis suppression followed by a decrease in ACTH release, and low levels of androgens and cortisol.
![]() Virilism results from the excessive secretion of androgens from the adrenal gland and often manifests as hirsutism in females.
Virilism results from the excessive secretion of androgens from the adrenal gland and often manifests as hirsutism in females.
The adrenal glands were first characterized by Eustachius in 1563. After Addison identified a case of adrenal insufficiency in humans, adrenal anatomy and physiology flourished. Most of the work done in the early and mid-1900s centered on the glucocorticoid cortisol. With the discovery of aldosterone by Simpson and Tait in 1952, adrenal pharmacology turned toward the mineralocorticoid. Conn1 followed with his classical description of primary aldosteronism (PA) in 1955, and numerous clinicians and investigators have continued to explore the variety of disease processes promoted through the adrenal gland.
PHYSIOLOGY, ANATOMY, AND BIOCHEMISTRY
The adrenal glands are located extraperitoneally to the upper poles of each kidney (Fig. 59-1). On average, each adrenal gland weighs 4 g and is 2 to 3 cm in width and 4 to 6 cm in length. The gland is fed by small arteries from the abdominal aorta and renal and phrenic arteries. Drainage of the adrenal gland occurs via the renal vein on the left and the inferior vena cava on the right.
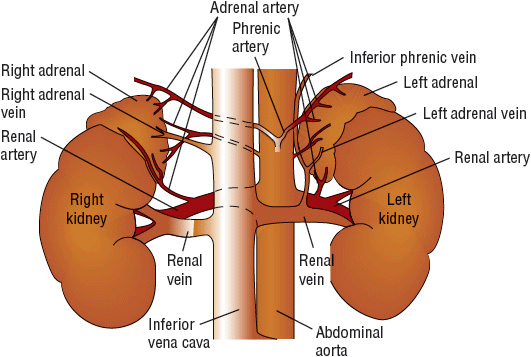
FIGURE 59-1 Anatomy of the adrenal gland.
The adrenal medulla occupies 10% of the total gland and is responsible for the secretion of catecholamines. The adrenal cortex accounts for the remaining 90% and is responsible for the secretion of three types of hormones (Fig. 59-2) from three separate zones.
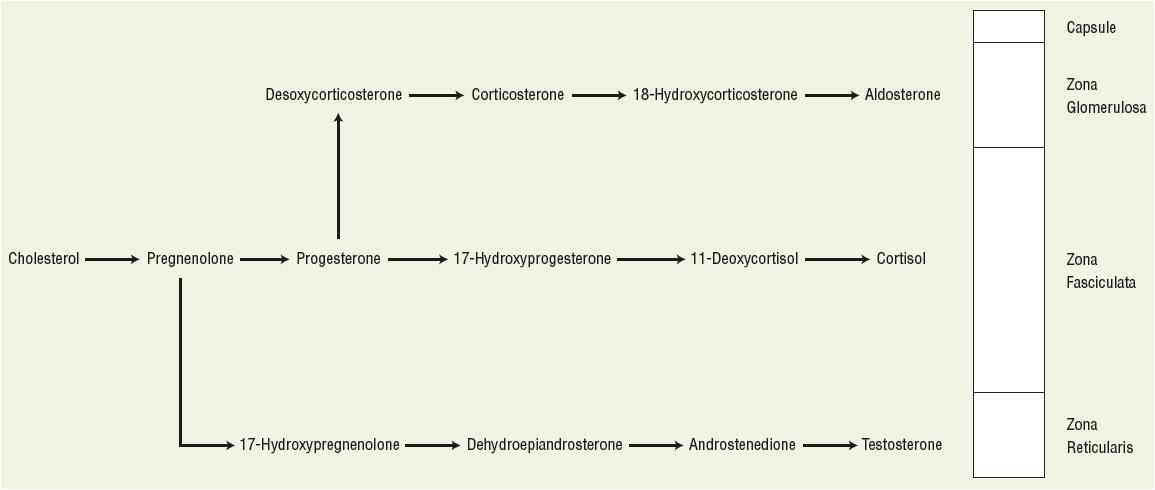
FIGURE 59-2 Hormone synthetic pathways in relation to the zones of the adrenal cortex.
The zona glomerulosa accounts for 15% of the total adrenal cortex and is responsible for mineralocorticoid production, of which aldosterone is the principal end product. Aldosterone maintains electrolyte and volume homeostasis by altering potassium and magnesium secretion and renal tubular sodium reabsorption. The zona fasciculata, the middle zone, makes up 60% of the cortex, is high in cholesterol, and is responsible for basal and stimulated glucocorticoid production. Glucocorticoids, mainly cortisol, are responsible for the regulation of fat, carbohydrate, and protein metabolism. The zona reticularis occupies 25% of the adrenal cortex, and is responsible for adrenal androgen production. The androgens, testosterone and estradiol, are the major end products and influence the reproductive system in addition to modulating primary and secondary sex characteristics.
Hormone Production and Metabolism
Adrenal steroid hormone synthesis begins with the conversion of cholesterol to pregnenolone by cytochrome P450 (CYP) enzymatic side-chain cleavage. Following this rate-limiting step, pregnenolone is converted to various 19- and 21-carbon steroids, depending on the enzymatic capabilities within each zone of the cortex. Androgenic properties predominate in the 19-carbon steroids, whereas mineralocorticoid and glucocorticoid properties manifest in the 21-carbon steroids.
Aldosterone production is initiated by the 21-hydroxylation of progesterone to form deoxycorticosterone. Subsequently, aldosterone synthase converts deoxycorticosterone to aldosterone through the intermediary, corticosterone. The zona glomerulosa preferentially produces aldosterone for three main reasons. First, the zona glomerulosa lacks 17α-hydroxylase activity and therefore can only convert pregnenolone to progesterone. Second, in contrast to the other zones, cells in the zona glomerulosa possess aldosterone synthase activity, which catalyzes the terminal steps in aldosterone synthesis. Lastly, cells of the zona glomerulosa display a greater number of angiotensin II receptors than cells of the other zones. Binding of angiotensin II to these receptors provides the stimulus for initiating the aldosterone biosynthesis cascade. Thus, aldosterone synthesis is a unique feature of the zona glomerulosa, explaining why aldosterone is not affected during disease processes limited to the zona fasciculata and/or reticularis.
Cortisol is produced from pregnenolone via four successive hydroxylations. These hydroxylations occur primarily in the zona fasciculata, although the zona reticularis is also capable of producing glucocorticoids.
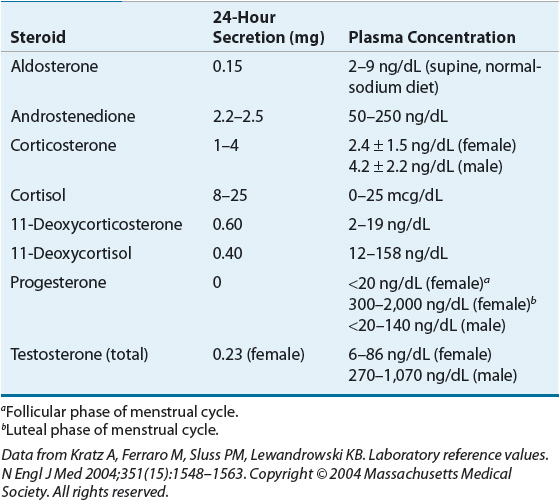
Androgens, produced primarily in the zona reticularis and less commonly in the zona fasciculata, have a 19-carbon structure and serve as precursors to more potent analogues produced in the periphery. The adrenal gland can synthesize estradiol and estrone from testosterone and androstenedione, respectively; however, these synthesized quantities are extremely small. The rates of production for the various steroids produced by the adrenal gland are listed in Table 59-1.
TABLE 59-1 Rates of Adrenal Production and Plasma Concentrations of Various Steroids
Glucocorticoid metabolism occurs in the liver and is responsible for converting inactive steroids to active metabolites, as well as modifying active steroids to less active or inactive metabolites. Most pharmaceutical steroid products are active; however, in the case of prednisone and cortisone, metabolism is necessary for conversion to the active prednisolone and cortisol, respectively.
Following metabolic conversion, glomerular filtration is primarily responsible for eliminating endogenously produced glucocorticoids. The half-life of cortisol is 70 to 120 minutes, whereas aldosterone exhibits extremely high intrinsic clearance and a corresponding half-life of only 15 minutes.
Metabolism and conversion of the various steroids can be altered by a variety of disease states and medicinal compounds. Drugs known to enhance steroid clearance include phenytoin, phenobarbital, rifampin, mitotane, and aminoglutethimide. Likewise, diseases such as hyperthyroidism and renal disease (dexamethasone only) can enhance steroid clearance. In contrast, drugs such as estrogens and estrogen-containing oral contraceptives reduce steroid clearance. Similarly, liver disease, age, pregnancy, hypothyroidism, anorexia nervosa, protein–calorie malnutrition, and renal disease (prednisolone only) are associated with reduced steroid clearance.
Plasma glucocorticoids are bound to one of three plasma proteins in varying degrees. Corticosteroid-binding globulin (CBG), albumin, and α1-glycoprotein are capable of binding glucocorticoids, with CBG being the principal binding protein. Steroid binding serves as a reservoir for steroids in their inactive state and ≥95% of cortisol is normally bound in this fashion. This binding prevents glucocorticoid activity at receptor-activating sites. Therefore, a final but important variable in altered plasma concentration of free (active) steroids is concentration of plasma proteins.
Regulation of Hormone Secretion
![]() Glucocorticoid secretion is regulated by the pituitary hormone, adrenocorticotropic hormone (ACTH [also known as corticotropin]). Under normal conditions, ACTH is released from the anterior pituitary in response to corticotropin-releasing hormone (CRH), which is secreted by the median eminence of the hypothalamus (Fig. 59-3). Vasopressin and oxytocin have weak ACTH-releasing activity through binding to the inferior V3 receptor. CRH, in combination with vasopressin and oxytocin, stimulates greater ACTH secretion than each hormone individually.
Glucocorticoid secretion is regulated by the pituitary hormone, adrenocorticotropic hormone (ACTH [also known as corticotropin]). Under normal conditions, ACTH is released from the anterior pituitary in response to corticotropin-releasing hormone (CRH), which is secreted by the median eminence of the hypothalamus (Fig. 59-3). Vasopressin and oxytocin have weak ACTH-releasing activity through binding to the inferior V3 receptor. CRH, in combination with vasopressin and oxytocin, stimulates greater ACTH secretion than each hormone individually.
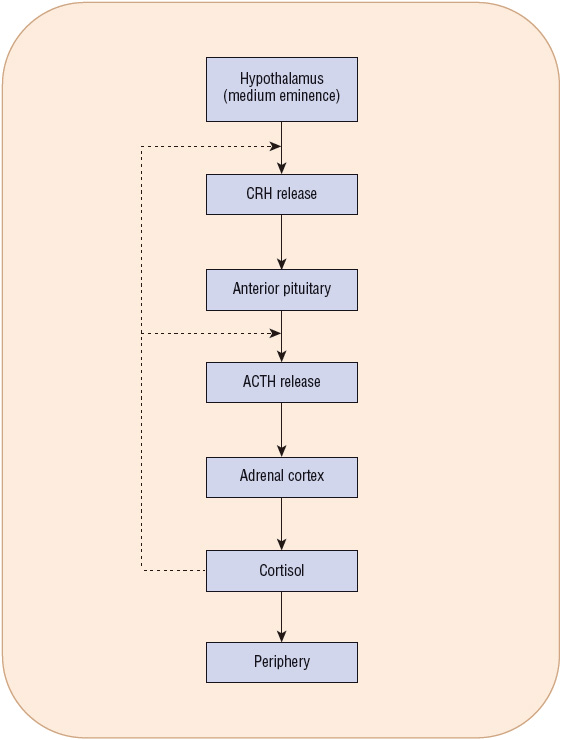
FIGURE 59-3 Negative feedback system involved in the regulation of cortisol secretion under normal conditions. (ACTH, adrenocorticotropic hormone; CRH, corticotropin-releasing hormone.)
Additionally, histochemical studies have demonstrated that certain neurotransmitters can stimulate production of CRH or ACTH directly. Specifically, both serotonin and norepinephrine have been shown to increase levels of ACTH. After release, ACTH stimulates the adrenal gland to release cortisol and, to a lesser extent, aldosterone and androgens. The rising cortisol concentration inhibits the secretion of CRH and ACTH through a negative feedback mechanism. In addition, leptin, an adipocyte hormone, can have an inhibitory effect on hypothalamic–pituitary–adrenal (HPA) activity.
Adrenal androgens are regulated in a similar fashion to cortisol. When plasma androgen reaches sufficient concentrations, production is terminated via a negative feedback loop. Androgen release is increased during puberty and in women with hirsutism. Adrenal androgen release decreases with age and in fasting states, including anorexia nervosa.
In contrast to cortisol and adrenal androgens, regulation of aldosterone secretion is considerably more complex. The renin–angiotensin system regulates aldosterone secretion through both intrarenal and extrarenal mechanisms. Renin production and subsequent aldosterone secretion is stimulated by blood pressure lowering (due to volume depletion), erect posture, salt depletion, β-adrenergic stimulation, and CNS excitation (see Chap. 15). Renin production is inhibited by salt loading, angiotensin II, vasopressin, potassium, calcium, blood pressure increases, and a variety of drugs. The renin-mediated production of angiotensin II is the initial stimulus for aldosterone synthesis. Additionally, angiotensin II can be acted on by aminopeptidase and converted to angiotensin III. Both angiotensin II and III are capable of stimulating the zona glomerulosa to secrete aldosterone. Following aldosterone secretion, increases in renal sodium and water retention as well as blood pressure occur, thereby turning off the stimulus for renin release.
HYPERFUNCTION OF THE ADRENAL GLAND
Adrenal disorders can be categorized as hyperfunction or hypofunction of the adrenal gland. Hyperfunction of the adrenal gland generally involves excess production of adrenal hormones, most notably cortisol, resulting in Cushing’s syndrome, or aldosterone, resulting in hyperaldosteronism.
Cushing’s Syndrome
In 1932, Cushing first described a syndrome of pituitary basophilism that attracted national attention. Until this time, no definitive diagnosis existed for patients with unexplained central obesity, cutaneous striae, osteoporosis, weakness, hypertension, diabetes mellitus, and congestion. Cushing emphasized that the disease was of a pituitary origin. Ten years later, Albright focused his attention on the sugar hormone, which he believed originated from the adrenal cortex.2
After the development of a method for measuring urinary steroids, Daughaday discovered elevated steroids in the urine of patients with Cushing’s syndrome. Finally, the end product was identified, and Cushing’s syndrome was correctly explained as an excess of cortisol in the plasma (hypercortisolism).
Etiology
Cushing’s syndrome results from the effects of supraphysiologic levels of glucocorticoids originating either from exogenous administration or, less commonly, from endogenous overproduction by the adrenal glands. Excess glucocorticoids are produced in response to overproduction of ACTH (ACTH-dependent) or by abnormal adrenocortical tissues regardless of ACTH stimulation (ACTH-independent). ACTH-dependent Cushing’s syndrome (≈80% of all Cushing’s syndrome cases) usually originates from overproduction of ACTH by the pituitary gland, which chronically stimulates the adrenal glands causing bilateral adrenal hyperplasia (BAH). Approximately 85% of these cases are caused by pituitary adenomas (Cushing’s disease). Ectopic ACTH-secreting tumors and nonneoplastic corticotropin hypersecretion, possibly secondary to excess CRH production, account for the remainder of ACTH-dependent causes.3 Ectopic ACTH syndrome refers to excessive ACTH production resulting from an endocrine or nonendocrine tumor, usually of the pancreas, thyroid, or lung. Small-cell carcinoma of the lung will lead to ectopic ACTH secretion in 0.5% to 2% of cases, whereas bronchial carcinoid tumors are usually the most common.4 Distinguishing between the various etiologies requires a careful history and pertinent laboratory work (Table 59-2).
TABLE 59-2 Various Etiologies of Cushing’s Syndrome and Their Respective Differences

The remaining 20% of Cushing’s syndrome cases are ACTH-independent and divided almost equally between adrenal adenomas and adrenal carcinomas, with rare cases caused by macronodular hyperplasia, primary pigmented nodular adrenal disease, and McCune-Albright syndrome.3,5 The majority of adrenal cortex tumors are benign adenomas. Adrenal carcinoma is found more often in children than in adults with Cushing’s syndrome.
Clinical Presentation
Patients with Cushing’s syndrome commonly present (>90% of patients) with central obesity and facial rounding. In addition, approximately 50% of patients will exhibit some peripheral obesity and fat accumulation. Fat accumulation in the dorsocervical area (buffalo hump) can be associated with any major weight gain, whereas increased supraclavicular fat pads are more specific for Cushing’s syndrome. Striae usually are present along the lower abdomen and take on a red to purple color. Traditionally, hypertensive complications have been major contributors to the morbidity and mortality of Cushing’s syndrome. Hypertension is diagnosed in 75% to 85% of patients, with diastolic blood pressures greater than 119 mm Hg noted in over 20% of patients.6 In addition, glucose intolerance is present in 60% of patients. Thus, many patients meet diagnostic criteria for the metabolic syndrome and have a corresponding increased risk of coronary heart disease (CHD) and stroke. Screening for Cushing’s syndrome in this population and in patients with uncontrolled diabetes mellitus has been suggested,7,8 particularly when these conditions surface at an unusually early age.9 However, screening all patients with type 2 diabetes is likely not cost-effective.10
Diagnosis
![]() The diagnosis of Cushing’s syndrome involves two steps: (a) establishing the presence of hypercortisolism, which is relatively easy, and (b) differentiating between etiologies, which can be challenging (Fig. 59-4).5,8,11 The presence of hypercortisolism can be established via the following tests: 24-hour UFC, midnight plasma cortisol, late-night salivary cortisol, and/or the low-dose DST (using 1 mg dexamethasone for the overnight test or 0.5 mg/6 hours for the classic 2-day study). However, because these tests cannot determine the etiology of Cushing’s syndrome, other tests and procedures will be subsequently employed. Such tests can include any of the following: plasma ACTH via immunoradiometric assay (IRMA) or radioimmunoassay (RIA); adrenal vein catheterization; metyrapone stimulation test; adrenal, chest, or abdominal computed tomography (CT); CRH stimulation test; IPSS; jugular venous sampling (JVS); cavernous sinus sampling; and pituitary magnetic resonance imaging (MRI). High-dose DST has been used in the past, but is no longer recommended due to its poor specificity and limited diagnostic value. Other possible tests and procedures include insulin-induced hypoglycemia, somatostatin receptor scintigraphy, the desmopressin stimulation test, the naloxone CRH stimulation test, the loperamide test, the hexarelin stimulation test, and radionuclide imaging.5,6,8,11–16 Table 59-3 summarizes some of the tests used to diagnose Cushing’s syndrome.
The diagnosis of Cushing’s syndrome involves two steps: (a) establishing the presence of hypercortisolism, which is relatively easy, and (b) differentiating between etiologies, which can be challenging (Fig. 59-4).5,8,11 The presence of hypercortisolism can be established via the following tests: 24-hour UFC, midnight plasma cortisol, late-night salivary cortisol, and/or the low-dose DST (using 1 mg dexamethasone for the overnight test or 0.5 mg/6 hours for the classic 2-day study). However, because these tests cannot determine the etiology of Cushing’s syndrome, other tests and procedures will be subsequently employed. Such tests can include any of the following: plasma ACTH via immunoradiometric assay (IRMA) or radioimmunoassay (RIA); adrenal vein catheterization; metyrapone stimulation test; adrenal, chest, or abdominal computed tomography (CT); CRH stimulation test; IPSS; jugular venous sampling (JVS); cavernous sinus sampling; and pituitary magnetic resonance imaging (MRI). High-dose DST has been used in the past, but is no longer recommended due to its poor specificity and limited diagnostic value. Other possible tests and procedures include insulin-induced hypoglycemia, somatostatin receptor scintigraphy, the desmopressin stimulation test, the naloxone CRH stimulation test, the loperamide test, the hexarelin stimulation test, and radionuclide imaging.5,6,8,11–16 Table 59-3 summarizes some of the tests used to diagnose Cushing’s syndrome.
TABLE 59-3 Summary of Tests Used to Diagnose Cushing’s Syndrome
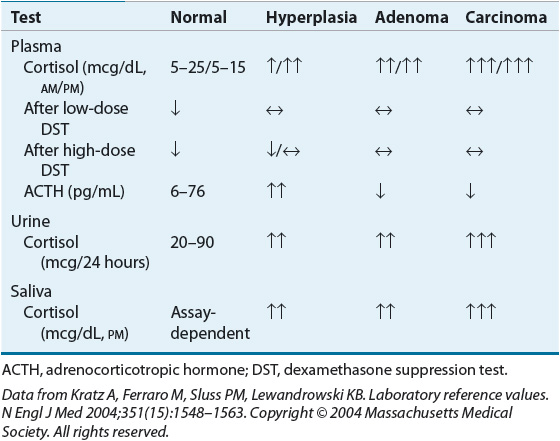
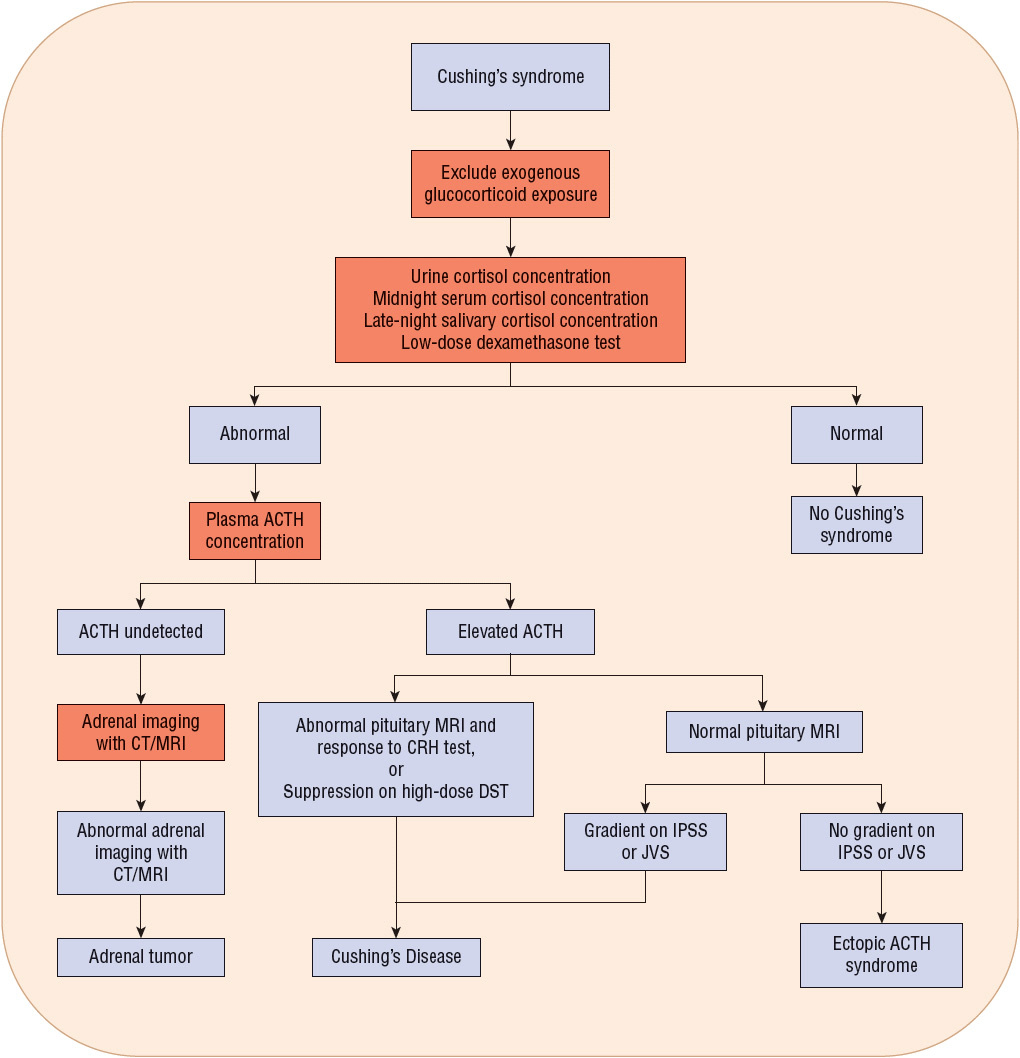
FIGURE 59-4 Algorithm for diagnosing Cushing’s syndrome. (ACTH, adrenocorticotropic hormone.)
CLINICAL PRESENTATION Cushing’s Syndrome
Elevated UFC concentrations are highly suggestive of Cushing’s syndrome, especially values fourfold greater than the upper limit of normal.3,13 In contrast to plasma measurements of cortisol, UFC measures only unbound cortisol. Consequently, the UFC test is unaffected by conditions and medications that alter CBG levels. Normal reference values for UFC are 20 to 90 mcg per 24-hour period. A twofold to threefold increase in urine cortisol is not uncommon in the patient with hyperfunction of the adrenal gland. Starvation, hydration from water loading (greater than 5 L/day), alcoholism, and acute stress are all capable of elevating urine cortisol concentrations. Likewise, elevated UFC results can occur during therapy with topical steroids, carbamazepine, and fenofibrate depending on the type of UFC test. Conversely, renal impairment (creatinine clearance [CrCl] of less than 60 mL/min) can falsely lower UFC concentrations. Because other pathologic conditions can increase the amount of free cortisol, additional tests may be warranted to confirm the diagnosis, or the diagnostic evaluation should be repeated when the acute stress has resolved. Of all urinary measures, UFC is the most useful assessment for patients with suspected Cushing’s syndrome.8,13,15
In healthy individuals, cortisol release follows a circadian rhythm whereby serum cortisol levels peak around 8:00 AM and thereafter decline by 60% to 80%, reaching a nadir between 3:00 and 4:00 AM. This rhythm is lost in the Cushing’s syndrome patient. Although many patients with Cushing’s syndrome will have serum cortisol values in the high normal range if the serum is assayed in the morning, only 3.4% will have normal values if measured late at night.17 Thus, a midnight serum cortisol greater than 7.5 mcg/dL (>1.8 mcg/dL if the patient is sleeping) is a highly sensitive assay for Cushing’s syndrome. However, this test is cumbersome and rarely recommended because it requires that patients be admitted for more than 48 hours to avoid false-positive responses secondary to the stress of hospitalization. An alternative assay is the measurement of late-night salivary cortisol. Salivary cortisol is highly correlated with free serum cortisol and independent of salivary flow rates. Moreover, salivary cortisol concentrations reflect changes in serum cortisol within minutes. Salivary cortisol can be considered as an acceptable alternative to UFC because of its convenience, stability (1 week), accuracy, and reproducibility. Unfortunately, normal reference ranges are assay-dependent, and cutoff points vary among institutions.18,19
In the overnight DST, 1 mg of dexamethasone is administered at 11:00 PM. The following morning at 8:00 AM fasting plasma cortisol is obtained for analysis. This supraphysiologic dose suppresses ACTH stimulation and cortisol production in healthy individuals. In contrast, the negative feedback loop is ineffective in patients with Cushing’s syndrome who generally exhibit morning cortisol concentrations above 5 mcg/dL. Some patients with Cushing’s syndrome administered the overnight DST can slightly suppress cortisol and using 1.8 mcg/dL as a cutoff can increase sensitivity, but at the expense of reduced specificity.20 Therefore, the overnight DST is useful only as a screening tool for Cushing’s syndrome. Drugs that induce or inhibit CYP3A4 metabolism can significantly alter dexamethasone levels, increasing the number of false-positive and false-negative DST tests. Concurrent measurements of dexamethasone levels with cortisol may improve the accuracy of testing for patients on CYP3A4-modifying drugs, although dexamethasone assays are not widely available. Also noteworthy, pregnancy and estrogen use (including oral contraceptives) increase CBG levels and frequently elicit false-positive results.13 Consequently, UFC testing is preferred over DST in these patient populations.
The first test used to determine the etiology of Cushing’s syndrome is the plasma ACTH test. Plasma ACTH concentrations can be measured via RIA or IRMA.12 In ACTH-dependent Cushing’s syndrome, ACTH can be normal or elevated. Very high levels of ACTH favor ectopic production. In contrast, ACTH values generally are low (less than 5 pg/mL) in ACTH-independent (adrenal) Cushing’s syndrome. Furthermore, ACTH levels can appear artificially low in some ectopic ACTH-producing tumors because ACTH can be secreted as an active prohormone that is not detected by the assay.
IPSS offers the highest sensitivity and specificity of any test in differentiating the etiology of Cushing’s syndrome. This technique requires catheterization of both petrosal sinuses with serial measurements of ACTH in each sinus and a peripheral vein after administration of CRH. A central-to-peripheral ACTH gradient is diagnostic for Cushing’s disease, whereas no gradient indicates ectopic ACTH production. Complications, such as venous thromboembolism, brain stem vascular damage, cost, and technical expertise, can limit use of this test.12 JVS uses the same concept as IPSS, is less invasive, and produces fewer complications; however, sensitivity is compromised.
Abnormal adrenal anatomy is effectively identified using high-resolution CT scanning and MRI.21 Nodules as small as 1 to 1.5 cm on the adrenal cortex are easily identified by CT. With the use of thin-section scanning, nodules as small as 3 to 5 mm can be visualized.22 Importantly, adrenal incidentalomas are prevalent in 5% to 10% of the general population. These masses may be functional (secreting), requiring intervention, or nonfunctional (nonsecreting), requiring only periodic observation. For this reason, abnormal imaging results are unable to conclusively diagnose adrenal disease when used alone. Nonadrenal imaging studies may be useful for identifying ectopic sources of ACTH secretion in patients for whom IPSS has ruled out Cushing’s disease.
Differential Diagnosis
Iatrogenic (exogenous) Cushing’s syndrome is the most common form of the disease. Therefore, all patients exhibiting hypercortisolism should undergo a comprehensive history and evaluation assessing medication use before laboratory testing is performed to identify endogenous causes. Iatrogenic Cushing’s syndrome can occur from administration of oral, inhaled, intranasal, intraarticular, and topical glucocorticoids, as well as progestins such as medroxyprogesterone acetate and megestrol acetate.23 Disease severity correlates with exogenous glucocorticoid potency, dose, frequency, route, and treatment duration. Moreover, patients taking CYP3A4 inhibitors concomitantly with a glucocorticoid can be at higher risk of developing iatrogenic Cushing’s syndrome.24,25 If exogenous glucocorticoids are being taken, plasma cortisol levels can increase, while corticosterone levels remain low.17
In the absence of any known exogenous causes, the clinician will need to differentiate the syndrome from other syndromes, such as pseudo-Cushing’s syndrome, that mimic Cushing’s. Patients with obesity, chronic alcoholism, depression, and acute illness of any type can present with certain features of Cushing’s syndrome. However, these patients may lack true Cushing’s syndrome. For example, depressed patients, although mimicking the urinary steroid abnormalities of Cushing’s syndrome, will not resemble a cushingoid patient in appearance. In chronic alcoholics, steroid laboratory panels generally return to baseline after ceasing alcohol intake. And obese patients often will have normal cortisol concentrations on both serum and urinary screening. Thus, identifying true cases of Cushing’s syndrome requires a comprehensive history in combination with laboratory and possibly imaging assessment.
TREATMENT
Desired Outcomes
![]() If left untreated, Cushing’s syndrome is associated with high morbidity and mortality due to associated disorders such as hypertension, diabetes mellitus, cardiovascular disease, and electrolyte abnormalities. These disorders limit the survival of the patient with Cushing’s syndrome to 4 to 5 years following initial diagnosis. The desired outcomes of treatment are to limit such detrimental outcomes and return the patient to a normal functional state by removing the source of hypercortisolism while minimizing pituitary or adrenal deficiencies.
If left untreated, Cushing’s syndrome is associated with high morbidity and mortality due to associated disorders such as hypertension, diabetes mellitus, cardiovascular disease, and electrolyte abnormalities. These disorders limit the survival of the patient with Cushing’s syndrome to 4 to 5 years following initial diagnosis. The desired outcomes of treatment are to limit such detrimental outcomes and return the patient to a normal functional state by removing the source of hypercortisolism while minimizing pituitary or adrenal deficiencies.
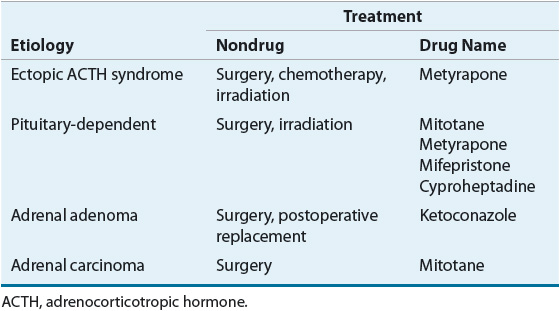
![]() The treatment of choice for both ACTH-dependent and ACTH-independent Cushing’s syndrome is surgical resection of any offending tumors.3,11 However, several secondary pharmacologic treatment plans are available, depending on the etiology of the disease (Table 59-4).26,27 These pharmacologic options are generally used in preoperative patients or as adjunctive therapy in postoperative patients awaiting response. Rarely, monotherapy is used as a palliative treatment when surgery is not indicated.
The treatment of choice for both ACTH-dependent and ACTH-independent Cushing’s syndrome is surgical resection of any offending tumors.3,11 However, several secondary pharmacologic treatment plans are available, depending on the etiology of the disease (Table 59-4).26,27 These pharmacologic options are generally used in preoperative patients or as adjunctive therapy in postoperative patients awaiting response. Rarely, monotherapy is used as a palliative treatment when surgery is not indicated.
TABLE 59-4 Possible Treatment Options in Cushing’s Syndrome Based on Etiology
Pharmacologic Therapy
![]() Pharmacotherapy of Cushing’s syndrome (dosing and monitoring parameters can be found in Tables 59-5 and 59-6, respectively)28 can be divided into four categories based on the anatomic site of action of the agent: (a) steroidogenesis inhibitors, (b) adrenolytic agents, (c) neuromodulators of ACTH release, and (d) glucocorticoid-receptor blocking agents.26,27
Pharmacotherapy of Cushing’s syndrome (dosing and monitoring parameters can be found in Tables 59-5 and 59-6, respectively)28 can be divided into four categories based on the anatomic site of action of the agent: (a) steroidogenesis inhibitors, (b) adrenolytic agents, (c) neuromodulators of ACTH release, and (d) glucocorticoid-receptor blocking agents.26,27
TABLE 59-5 Drug Dosing in the Treatment of Cushing’s Syndrome
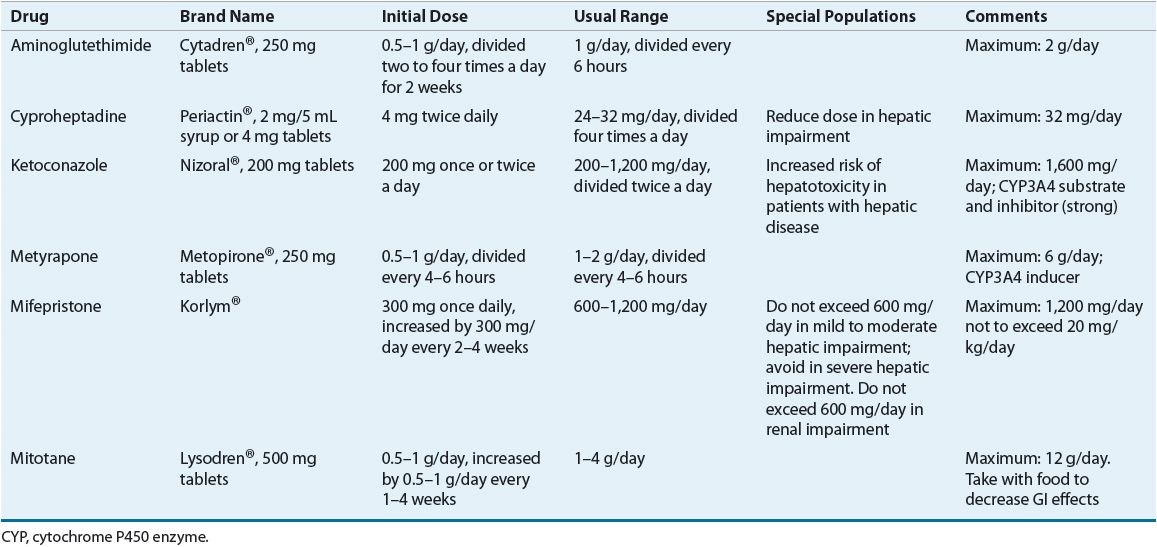
TABLE 59-6 Drug Monitoring in the Treatment of Cushing’s Syndrome
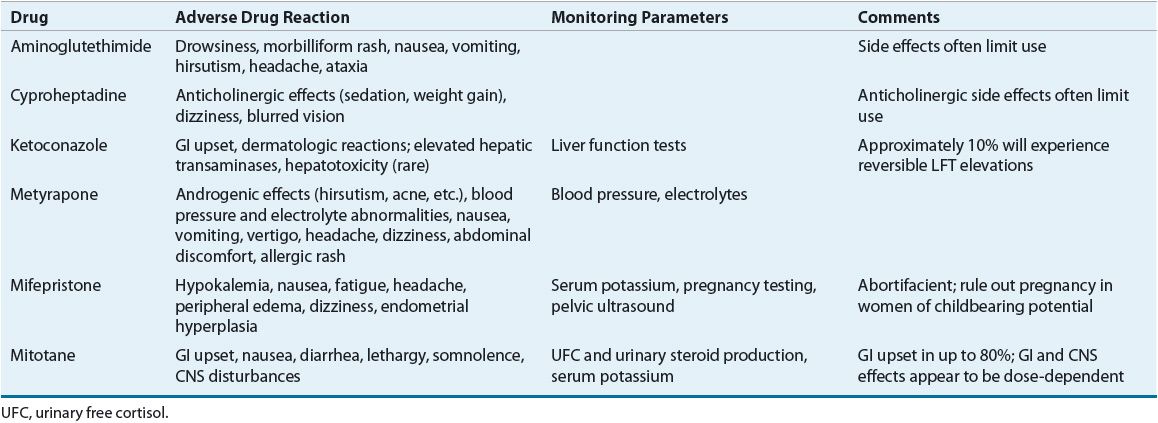
Steroidogenesis Inhibitors
As their name implies, steroidogenesis inhibitors block the production of cortisol. This class includes metyrapone, ketoconazole, etomidate, and aminoglutethimide. Metyrapone inhibits 11β-hydroxylase, the enzyme responsible for converting 11-deoxycortisol to cortisol. Following administration, a sudden decrease in cortisol levels occurs within hours and prompts a compensatory rise in plasma ACTH concentrations. As ACTH increases and blockage of cortisol synthesis persists, adrenal steroidogenesis efforts are shunted toward androgen production. Consequently, metyrapone is associated with significant androgenic side effects, including hirsutism and increased acne, making it less ideal for females. In addition, metyrapone blocks aldosterone synthesis and causes the accumulation of aldosterone precursors, which exhibit weak mineralocorticoid activity. Blood pressure and electrolyte level variations can ensue, depending on the level of circulating 11-deoxycortisol and the degree of aldosterone inhibition. Additional adverse effects, including nausea, vomiting, vertigo, headache, dizziness, abdominal discomfort, and allergic rash, have been reported following administration, but are often signs of overtreatment.26,27,29 Metyrapone is currently available through the manufacturer only for compassionate use.
The imidazole derivative antifungal, ketoconazole, effectively inhibits steroidogenesis via multiple mechanisms when used in large doses. In contrast to the quick onset of metyrapone, the benefits of ketoconazole therapy are achieved only after several weeks of therapy. In addition to lowering serum cortisol levels, ketoconazole exhibits antiandrogenic activity attributable to its inhibition of multiple CYP enzymes as well as 11β-hydroxylase and 17α-hydroxylase.26 This activity may be beneficial in female patients with Cushing’s syndrome, but can cause gynecomastia and hypogonadism in males. Sustained therapy with ketoconazole also imparts beneficial effects on serum cholesterol profiles, including lowering total and LDL cholesterol levels. Ketoconazole induces a reversible elevation of hepatic transaminases in approximately 10% of patients.30 Frequent liver function monitoring is recommended, although progression to serious hepatotoxicity is rare. Additional common adverse effects include GI discomfort and dermatologic reactions.
Ketoconazole may be used concomitantly with metyrapone to achieve synergistic reductions in cortisol levels. Because these drugs differ in their onset of action, coadministration allows for more complete suppression of cortisol synthesis. Moreover, the antiandrogenic actions of ketoconazole therapy may offset the androgenic potential of metyrapone, thus attenuating a major limitation of metyrapone therapy.
The anesthetic etomidate is an imidazole derivative similar to ketoconazole that inhibits 11β-hydroxylase.26 Etomidate is available only in a parenteral formulation and is therefore limited to patients with acute hypercortisolemia requiring emergency treatment.
Initially, aminoglutethimide was used to treat refractory forms of epilepsy, but was later discovered to be a potent inhibitor of adrenal steroid synthesis. Aminoglutethimide inhibits the conversion of cholesterol to pregnenolone early in the steroid biosynthesis pathway, thereby inhibiting the production of cortisol, aldosterone, and androgens. Owing to these broad inhibitory actions, side effects, including severe sedation, nausea, ataxia, and skin rashes, limit the use of aminoglutethimide in many patients.26,27 Moreover, because other steroidogenesis inhibitors offer greater efficacy combined with fewer side effects, aminoglutethimide has fallen out of favor in the treatment of Cushing’s syndrome. If aminoglutethimide is used, it should be coadministered with another steroidogenesis inhibitor, usually metyrapone, secondary to high relapse rates with aminoglutethimide monotherapy.
Adrenolytic Agents
Mitotane is a cytotoxic drug that structurally resembles the insecticide dichlorodiphenyltrichloroethane (DDT). Mitotane inhibits the 11-hydroxylation of 11-desoxycortisol and 11-desoxycorticosterone in the cortex, resulting in a net inhibition of cortisol and corticosterone synthesis. Similar to ketoconazole, mitotane takes weeks to months to exert beneficial effects. Sustained cortisol suppression occurs in most patients and may persist following discontinuation of therapy in up to one third of patients. Because of its cytotoxic nature, mitotane degenerates cells within the zona fasciculata and reticularis, resulting in atrophy of the adrenal cortex. The zona glomerulosa is minimally affected during acute therapy but can become damaged following long-term treatment.28,31
Importantly, mitotane can induce significant neurologic and GI side effects and patients should be monitored carefully or hospitalized when initiating therapy. Nausea and diarrhea are common adverse effects that occur at doses greater than 2 g/day and can be avoided by gradually increasing the dose and/or administering the agent with food. Approximately 80% of patients treated with mitotane develop lethargy and somnolence, and other CNS adverse drug reactions occur in approximately 40% of patients. Furthermore, significant but reversible hypercholesterolemia and prolongation of bleeding times can result from mitotane use.26,27 Mitotane increases production of CBG resulting in artifactually elevated plasma cortisol; thus, UFC and urinary steroid production should be monitored to assess response to therapy.26 If necessary, steroid replacement therapy can be given. However, because mitotane also increases extraadrenal metabolism of exogenously administered corticosteroids (especially hydrocortisone), higher steroid replacement doses may be required.
Neuromodulatory Agents
Pituitary secretion of ACTH is normally mediated by various neurotransmitters, including serotonin, GABA, acetylcholine, and the catecholamines. Although ACTH-secreting pituitary tumors (Cushing’s disease) self-regulate ACTH production to some degree, these neurotransmitters are still capable of promoting pituitary ACTH production. Consequently, agents that target these neurotransmitters have been proposed for the treatment of Cushing’s disease. Such agents include cyproheptadine, ritanserin, ketanserin, bromocriptine, cabergoline, valproic acid, octreotide, lanreotide, rosiglitazone, and tretinoin. However, none of these drugs have demonstrated consistent clinical efficacy in the treatment of Cushing’s disease.
Cyproheptadine, a nonselective serotonin receptor antagonist and anticholinergic drug, can decrease ACTH secretion in some Cushing’s disease patients. However, side effects, including sedation and weight gain, significantly limit the use of this drug. Likewise, selective serotonin type 2 receptor antagonists, including ritanserin and ketanserin, have demonstrated limited efficacy. Owing to their poor efficacy and high relapse rates, these drugs should be avoided except in nonsurgical candidates refractory to more conventional treatments.
Dopamine D2 receptor agonists, including bromocriptine and cabergoline, initially reduce ACTH secretion in as many as half of all patients with Cushing’s disease. This action occurs through activation of inhibitory D2 receptors that are expressed in approximately 80% of pituitary adenomas.32 Reductions in ACTH levels are often minor and rarely sustained with long-term bromocriptine therapy. Cabergoline exhibits a higher specificity and affinity for D2 receptors as well as a prolonged half-life compared with bromocriptine. These differences may explain the greater response rates observed with cabergoline monotherapy; however, a sustained response occurs in only 30% to 40% of patients.33,34
The somatostatin analogues, octreotide and lanreotide, generally are ineffective in reducing ACTH secretion in Cushing’s disease. These two agents primarily target somatostatin receptor subtype 2 (sst2), whereas pituitary adenomas predominantly express sst5. Pasireotide, a recently approved somatostatin analogue, exhibits a high affinity for sst1, sst2, sst3, and, especially, sst5 receptor subtypes. In a phase 3 study of 162 adults with Cushing’s disease and an elevated UFC level, pasireotide administered at 600 or 900 mcg injected subcutaneously twice daily reduced the median UFC by 50% by month 2; levels remained stable for the duration of the 12-month study. Clinical signs and symptoms of Cushing’s disease were also improved as were blood pressure, weight, LDL cholesterol, and quality of life. Side effects were mostly GI in nature, although 73% of subjects experienced an adverse event related to hyperglycemia; preexisting diabetes mellitus or impaired glucose tolerance increased the risk for these events. Notably, glycated hemoglobin A1c increased by an average of 1.4%. Gallstones were also rarely seen with six subjects undergoing cholecystectomy.35
Glucocorticoid-Receptor Blocking Agents
Mifepristone (RU-486) is a potent progesterone- and glucocorticoid-receptor antagonist that inhibits dexamethasone suppression and increases endogenous cortisol and ACTH levels in normal subjects.26,29 Clinical experience and trial data in Cushing’s syndrome suggest that RU-486 is highly effective in reversing the manifestation of hypercortisolism, including hyperglycemia, hypertension, and weight gain.36 However, because of its novel site of action, RU-486 induces a compensatory rise in ACTH and cortisol levels. Consequently, efficacy and toxicity monitoring must rely on clinical signs rather than laboratory assessments. Common adverse effects of RU-486 include fatigue, nausea, headache, arthralgia, peripheral edema, endometrial thickening (with or without vaginal bleeding), and significant reductions in serum potassium. Oral potassium supplementation or spironolactone can be effective in mitigating the latter adverse effect, although high doses may be required.36
Close monitoring of 24-hour UFC levels and serum cortisol levels is essential to detect treatment-induced adrenal insufficiency. Steroid secretion should be monitored with all of these drugs except RU-486 and steroid replacement given as needed. Whatever the choice, pharmacologic therapy in pituitary-dependent disease is mainly centered around patient stabilization prior to surgery or in patients waiting for potential response to other therapies.



