THE ADRENAL GLAND: AN OVERVIEW
The adrenal gland is a multifunctional organ that produces steroid hormones and neuropeptides essential for life. Despite the complex actions of adrenal hormones, most pathological conditions of the adrenal gland are manifested by their impact on blood pressure, electrolyte balance, and androgen excess.1 An adrenal etiology should be considered in the differential diagnosis when patients presents with (i) hypertension, hypokalemia, and metabolic alkalosis (suspect hyperaldosteronism); (ii) hypertension, spells of anxiety, palpitations, dizziness, and diaphoresis (pheochromocytoma); (iii) hypertension, rapid unexplained weight gain, red/purple stretch marks, and proximal muscle weakness (Cushing’s syndrome); (iv) inappropriate hirsutism/virilization and inability to conceive (congenital adrenal hyperplasia); and (v) loss of appetite, unintentional weight loss, and pigmented skin (primary adrenal insufficiency).
In clinical practice, patients often present with underproduction or overproduction of one or more adrenal hormones. Adrenal nodules are also discovered incidentally on abdominal imaging performed for other reasons. Hypofunction is treated with hormone replacement, and hyperfunction is treated with pharmacologic suppression and/or surgery.
EMBRYOLOGY AND ANATOMY
The adrenal gland is composed of two embryologically distinct tissue—the outer adrenal cortex and inner adrenal medulla. The cortex is derived from mesenchymal cells located near the urogenital ridge that differentiate into three structurally and functionally distinct zones (Fig. 21.1). The medulla arises from neural crest cells that invade the cortex during the 2nd month of fetal development. By adulthood, the medulla contributes 10% of total adrenal weight.
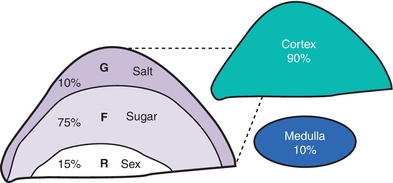
FIGURE 21.1 Adrenal gland by layer.
Adult adrenal glands are shaped like pyramids, located superior and medial to the upper pole of the kidneys in the retroperitoneal space (hence, also known as suprarenal glands). On cross section, both regions remain distinct; the cortex appears yellow, while the medulla is dark mahogany.2
Adrenal arterial supply is symmetric. Small arterioles branch to form a dense subcapsular plexus that drains into the sinusoidal plexus of the cortex. There is no direct arterial blood supply to middle and inner zones. In contrast, venous drainage from the central vein displays laterality. After crossing the medulla, the right adrenal vein empties into the inferior vena cava, and the left adrenal vein drains into the left renal vein. There is a separate capillary sinusoidal network from the medullary arterioles that also drains into the central vein and limits the exposure of cortical cells to medullary venous blood. Glucocorticoids from the cortex are carried directly to the adrenal medulla via the portal system, where they stimulate enzyme production of epinephrine (EPI) (Fig. 21.15).
Sympathetic and parasympathetic axons reach the medulla through the cortex. En route, these axons release neurotransmitters (e.g., catecholamines and neuropeptide Y) that modulate cortex blood flow, cell growth, and function. Medullary projections into the cortex have been found to contain cells that also synthesize and release neuropeptides, such as vasoactive inhibitory peptide (VIP), adrenomedullin, and atrial natriuretic peptide (ANP), and potentially influence cortex function.
THE ADRENAL CORTEX BY ZONE
The major adrenal cortical hormones, aldosterone, cortisol, and dehydroepiandrosterone sulfate (DHEA-S), are uniquely synthesized from a common precursor cholesterol by cells located in one of three functionally distinct zonal layers of the adrenal cortex. These zonal layers are zona glomerulosa, zona fasciculata, and zona reticularis, respectively (Fig. 21.1).
Zona glomerulosa (G-zone) cells (outer 10%) synthesize aldosterone, a mineralocorticoid critical for sodium retention, potassium excretion, acid–base homeostasis, and regulation of blood pressure. They have low cytoplasmic-to-nuclear ratios and small nuclei with dense chromatin with intermediate lipid inclusions.
Zona fasciculata (F-zone) cells (middle 75%) synthesize glucocorticoids, such as cortisol and cortisone, critical for glucose homeostasis and blood pressure. Fasciculata cells are cords of clear cells, with a high cytoplasmic-to-nuclear ratio and lipids laden with “foamy” cytoplasm. The fasciculata cells also generate androgen precursors such as dehydroepiandrosterone (DHEA), which is sulfated in the innermost zona reticularis (R-zone). Subcapsular adrenal cortex remnants can regenerate into fasciculate adrenals, metastasize, and survive in ectopic locations such as the liver, gallbladder wall, broad ligaments, celiac plexus, ovaries, scrotum, and cranium.
Zona reticularis cells (inner 15%) sulfate DHEA to DHEA-S, which is the main adrenal androgen. The zone is sharply demarcated with lipid-deficient cords of irregular, dense cells with lipofuscin deposits.
Adrenal cell types are presumed to arise from stem cells. A proposed tissue layer between the zona glomerulosa and fasciculata may serve as a site for progenitor cells to regenerate zonal cells.3
Cortex Steroidogenesis
Control of steroid hormone biosynthesis is complex, including adrenocorticotropic hormone (ACTH) and angiotensin II (AT II). It occurs via substrate availability, enzyme activity, and inhibitory feedback loops that are layer specific.
All adrenal steroids are derived by sequential enzymatic conversion of a common substrate, cholesterol. Adrenal parenchymal cells accumulate and store circulating low-density lipoproteins (LDLs). The adrenal gland can also synthesize additional cholesterol using the enzyme acetyl-CoA, ensuring that adrenal steroidogenesis remains normal in patients with variable lipid disorders and in patients on lipid-lowering agents.
Only free cholesterol can enter steroidogenic pathways in response to ACTH. The availability of free intracellular cholesterol is metabolically regulated by ACTH (stimulatory) and LDL (inhibitory) through multiple mechanisms. Corticotropin-releasing hormone (CRH) is secreted from the hypothalamus in response to circadian signals, low serum cortisol levels, and stress. CRH stimulates release of stored ACTH from the anterior pituitary gland, which stimulates transport of free cholesterol into adrenal mitochondria, initiating steroid production.
Conversion of cholesterol to pregnenolone is a rate-limiting step in steroid biosynthesis: six carbon atoms are removed from cholesterol by enzyme cytochrome P450 (CYP450) present in the mitochondrial membrane (Fig. 21.2). Newly synthesized pregnenolone is then returned to the cytosol for subsequent zonal conversion by microsomal enzymes in each layer by F-zone enzymes and/or androgens by enzymes in the R-zone (Fig. 21.3).
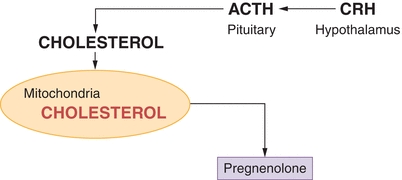
FIGURE 21.2 Conversion of cholesterol to pregnenolone. ACTH, adrenocorticotropic hormone; CRH, corticotropin-releasing hormone.
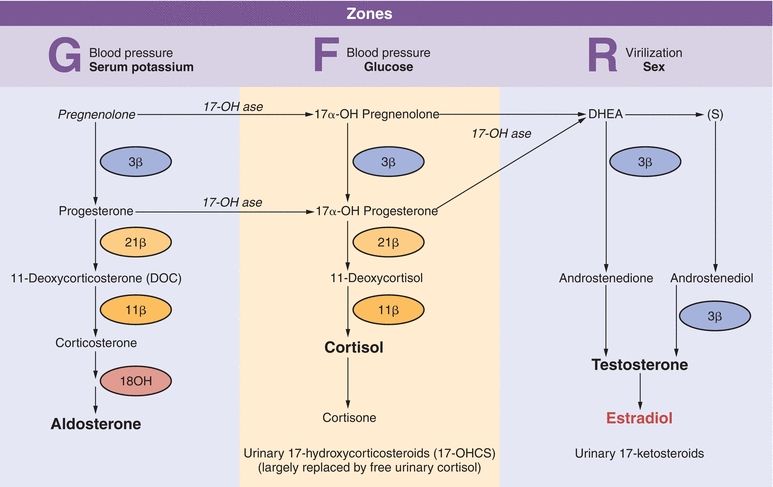
FIGURE 21.3 Adrenocortical hormone synthesis by zone.
High serum glucocorticoids suppress release of CRH and ACTH via a negative feedback mechanism. Cortisol is the primary feedback regulator of ACTH-stimulated hormone production in the adrenal cortex. ACTH generally does not impact G-zone aldosterone synthesis, although cortisol has mineralocorticoid action.
Decreased activity of any enzyme required for biosynthesis can occur as an acquired or inherited (autosomal recessive) trait. Defects that decrease the production of cortisol cause increases in ACTH and CRH secretion in an attempt to stimulate cortisol levels and lead to adrenal hyperplasia or overproduction of androgens, depending on the affected enzyme.
Evaluation of adrenal function requires measuring relevant adrenal hormones, metabolites, and regulatory secretagogues. Diagnosis is based on the correlation of clinical and laboratory findings.4
Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia refers to a group of clinical entities that arise from absent or diminished activity of enzymes involved in steroidogenesis. The mineralocorticoid, glucocorticoid, and androgen production pathways can be affected to varying degrees based on the enzyme affected. Blocks in one pathway result in upstream substrate buildup and potential upregulation of another pathway. The most common enzyme affected is 21-hydroxylase. Deficiency in this enzyme results in decreased glucocorticoid, in some cases mineralocorticoid and increased adrenal androgen production.5 A very high serum concentration of 17-hydroxyprogesterone, the normal substrate for 21-hydroxylase, is diagnostic of classic 21-hydroxylase deficiency. The “classic” presentation is seen in infants. It presents with features such as failure to thrive and low blood pressure. These infants need both glucocorticoid and mineralocorticoid replacement. A second “nonclassic” form is seen in adults. Woman with this form present in their reproductive years with complaints of hirsutism, menstrual irregularities, and infertility. They may need steroids during pregnancy. The different enzyme defects along with clinical and biochemcial abnormalities have been summarized in Figure 21.4.
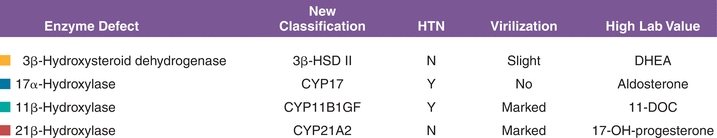
FIGURE 21.4 Congenital adrenal hyperplasia syndromes.
PRIMARY ALDOSTERONISM
Overview
The clinical entity in which excessive secretion of aldosterone cannot be suppressed with salt or volume replacement is termed primary aldosteronism (PA). PA results in hypertension, hypokalemia, metabolic alkalosis, and increased risk of vascular disease such as stroke as depicted in Figure 21.5. It is estimated that 5% to 10% of patients with hypertension have PA.6 Endocrine Society guidelines on PA suggest that hypokalemia is found only in 37% of patients with PA.7 The guidelines recommend more aggressive screening may be appropriate in those with SBP greater than 160/DBP greater than 100, drug-resistant hypertension, hypokalemia associated with hypertension, presence of adrenal mass, family history of early hypertension or stroke, and first-degree relatives of patients with PA.

FIGURE 21.5 G-zone function and pathology. HTN, hypertension.
Etiology
The most common causes of PA are:
- Aldosterone-producing adrenal adenoma
- Unilateral or bilateral adrenal hyperplasia
Other causes are as follows:
- Familial hyperaldosteronism such as glucocorticoid remediable aldosteronism (GRA)
- Adrenocortical carcinomas that secrete aldosterone
- Ectopic aldosterone secretion
Diagnosis
Plasma aldosterone concentration (PAC) and plasma renin activity (PRA) are used in the diagnosis of PA.8 Presence of both PAC of greater than 15 ng/dL and PAC/PRA (plasma renin activity) of 30 or greater is suggestive of PA. The 2008 Endocrine Society guidelines recommend the labs be checked in AM after the patient has been up for at least 2 hours. The patient should be seated for 5 to 15 minutes before the draw and sodium intake should be unrestricted. Most antihypertensive medications do not need to be stopped prior to testing except mineralocorticoid antagonist. If clinical suspicion is high in a patient on ACE-I and testing is negative, the ACE-I should be discontinued and testing repeated. If the initial screening is suggestive of PA, confirmatory tests include aldosterone measurement following either oral salt loading or IV saline infusion.
Oral salt loading requires oral consumption of about 5,000 mg NaCl per day. On day 3, urine sodium, creatinine, and aldosterone are measured in a 24-hour urine collection. Presence of urine sodium greater than 200 mEq and urine aldosterone greater than 12 μg/24 hours is suggestive of PA. The saline infusion test involves IV infusion of 2 L NaCl in 2 hours. Following this, PAC normally will be less than 5 ng/dL, and in those with PA, the value is often greater than 10 ng/dL.
Biochemical evaluation is followed by adrenal imaging (CT, MRI). Presence of an adrenal mass should still prompt further testing before surgical intervention since adrenal adenomas are often nonfunctional. Aldosterone secretion may be due to bilateral hyperplasia. Adrenal venous sampling (AVS) is recommended to distinguish between hyperplasia and aldosterone-secreting adenoma.9 Some authors do suggest that in persons less than 35 years, one may be able to proceed to surgery without AVS. The study requires technical expertise and should be performed by experienced practitioners. Adrenal vein to IVC cortisol ratios and aldosterone levels corrected for cortisol are used to determine appropriate placement of sampling catheters. If there is asymmetry in aldosterone release between adrenal glands (greater than 4:1), it is likely to be unilateral excess aldosterone production and the patient would benefit from surgical intervention. If less than 3:1 ratio exists, it is likely to be secondary to adrenal hyperplasia and the patient should be treated medically.
Treatment
Surgery is the treatment of choice for aldosterone-producing adenoma. Hypertension is controlled in 30% to 60% of the patients treated surgically.10 Mineralocorticoid antagonists such as spironolactone or eplerenone should be used for adrenal hyperplasia. If GRA is suspected in patients with multiple family members with early onset of hypertension, steroids such as prednisone are the treatment of choice.
Isolated Hypoaldosteronism
Insufficient aldosterone secretion is seen with adrenal gland destruction, with chronic heparin therapy, following unilateral adrenalectomy (transient), and with G-zone enzyme deficiencies. Most hypoaldosteronism occurs in patients with mild renal insufficiency such as persons with diabetes who present with mild metabolic acidosis, high serum potassium+, low urinary potassium+ excretion (urine K+ < urine Na+), and low renin. Treatment is with dietary changes and Fludrocortisone (a synthetic mineralocorticoid), which enhances salt retention and the secretion of potassium+ and hydrogen+.
Types of aldosteronism are diagrammed in Figure 21.6 according to their PA (y-axis) and PRA (x-axis) values.
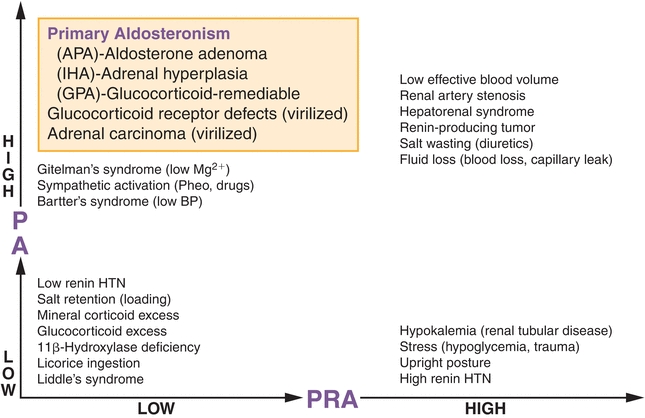
FIGURE 21.6 Types of aldosteronism according to plasma aldosterone:plasma renin activity (PRA) ratio. HTN, hypertension.
ADRENAL CORTICAL PHYSIOLOGY
Cortisol synthesis (8 to 15 mg/d) is critical to hemodynamics and glucose homeostasis. F-zone disorders manifest with blood pressure and glucose abnormalities (Fig. 21.7). Glucocorticoids maintain blood glucose by inducing lipolysis and causing amino acid release from muscle for conversion into glucose (gluconeogenesis) and storage as liver glycogen.
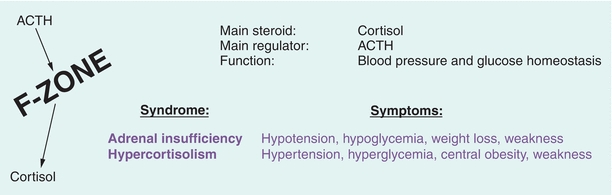
FIGURE 21.7 F-zone function and pathology. ACTH, adrenocorticotropic hormone.
ADRENAL INSUFFICIENCY
Overview
Adrenal insufficiency is a term that describes inadequate hormone secretion from the adrenal cortex. This may be a primary adrenal problem in which case there is inadequate release of glucocorticoids (such as cortisol), mineralocorticoids (such as aldosterone), and adrenal androgens. In primary adrenal insufficiency, there is reduced production of adrenal hormones despite adequate stimulation. In secondary adrenal insufficiency, adrenal gland function is preserved but the stimulus for hormone release is insufficient or absent. This would occur if the pituitary gland failed to release adequate adrenocorticotropic hormone (ACTH). The most common cause of adrenal insufficiency is autoimmune destruction of the adrenal gland.11 Other causes of adrenal insufficiency include infections (tuberculosis and histoplasmosis), tumors, bilateral adrenal hemorrhage, etc.
Symptoms
Clinical symptoms of chronic adrenal insufficiency can be nonspecific with most patients complaining of fatigue, decreased appetite, weight loss, and nausea. Patients may also present with more severe findings such as low blood pressure, low blood sugar, low serum sodium, and high potassium levels as depicted in Figure 21.8.

FIGURE 21.8 Signs and symptoms of adrenal insufficiency.
Diagnosis
The diagnosis of adrenal insufficiency can be suspected if the 8 am serum cortisol is low with concomitant elevation in ACTH levels. A cortisol level less than 3 μg/dL in the morning is highly suggestive of adrenal insufficiency. The diagnosis of primary adrenal insufficiency is made by performing an ACTH stimulation test. The test is performed at 8 am in the fasting state.12 Baseline cortisol and ACTH levels are obtained. The patient is then given 250 μg of cosyntropin (synthetic ACTH) intravenously and cortisol level is checked at 30 and 60 minutes post ACTH administration. A cortisol of 18 or greater at either 30 or 60 minutes post ACTH time point suggests normal adrenal function. In secondary adrenal insufficiency, the ACTH stimulation test may be normal or abnormal based on the duration of the disease. If secondary adrenal insufficiency is suspected, metyrapone suppression testing can be done.13 Metyrapone blocks certain enzymes in the steroidogenesis pathway. When metyrapone is administered orally at midnight, in normal individuals, it will block 11β-hydroxylase increasing 11-deoxycortisol (>7 μg/dL) while cortisol decreases (<5 μg/dL). Secondary adrenal insufficiency is suggested in patients with a near-normal response to a 250-μg cosyntropin test but with an abnormal response to metyrapone. An alternative is to do an insulin tolerance test (ITT). Hypoglycemia is induced with insulin administration and ACTH, cortisol, and growth hormone response to hypoglycemia is measured. The ITT should be avoided in those with cardiovascular disease and seizure disorder. Figure 21.9 provides a summary of primary and secondary adrenal insufficiency.
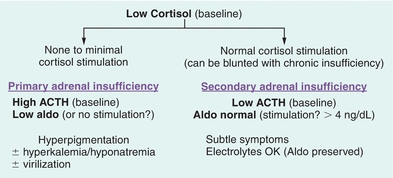
FIGURE 21.9 Differential diagnosis of low cortisol states. ACTH, adrenocorticotropic hormone.
Treatment
The treatment of primary adrenal insufficiency includes both glucocorticoid replacement (prednisone or hydrocortisone) and mineralocorticoid replacement (fludrocortisone). In secondary adrenal insufficiency, only glucocorticoid replacement is required since aldosterone production remains normal.
HYPERCORTISOLISM (CUSHING’S SYNDROME)
Overview
Excess cortisol production presents with a constellation of clinical manifestations. Each is not unique to cortisol excess but when present together may suggest hypercortisolism. The common findings are as follows: There is progressive central obesity with sparing of extremities. Dorsocervical fat pad deposition and fat accumulation in the cheeks result in the “buffalo hump” and “moon face,” respectively. A number of dermatologic manifestations can be seen such as atrophy of skin, easy bruising, wide purple striae, and hyperpigmentation based on the etiology of cortisol excess. In women, menstrual irregularity, signs of androgen excess such as acne, and hirsutism can be seen. Proximal muscle weakness and decreased bone mineral density may be present. Patients with cortisol excess also have increased risk of cardiovascular disease, thromboembolic events such as DVT, increased anxiety, and risk of infection including atypical fungal infections.14
Etiology
Cortisol excess may be due to adrenal tumors that make excess cortisol in which case pituitary release of ACTH levels is low. Alternatively, cortisol production could be due to elevated ACTH levels either from a pituitary tumor or from nonpituitary or ectopic sources often associated with malignancy such as lung cancer. The term Cushing’s disease is used when the source of elevated ACTH is the pituitary gland, while other forms of cortisol excess are labelled as Cushing’s syndrome. Cushing’s disease is the most common reason for cortisol excess accounting for about 70% of cases (Figs. 21.10 and 21.11).

FIGURE 21.10 Conditions associated with hypercortisolism. HTN, hypertension.
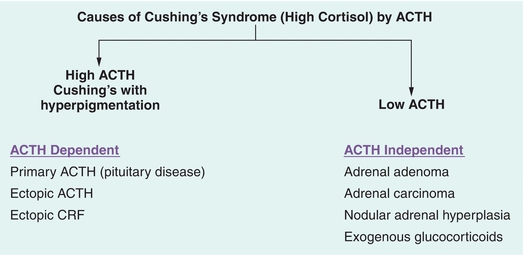
FIGURE 21.11 Differentiating source of adrenocorticotropic hormone (ACTH) secretion.
Diagnosis
Once exogenous cortisol use (oral, topical, injected, inhaled ) is ruled out, initial screen tests for cortisol excess include (a) urine free cortisol collected over 24 hours (on two occasions to be considered positive), (b) midnight salivary cortisol (two occasions), and (c) dexamethasone suppression testing (1 mg overnight or 2 mg over 48 hours). A systematic review during the development of Endocrine Society guidelines in 2008 suggested that the three tests were similar in diagnostic accuracy.15 The guidelines recommend demonstrating at least two positive tests for the diagnosis as this increases the diagnostic accuracy over any one test.
Urine free cortisol (UFC) measurement requires accurate collection and measurement of urine volume. Fluid intake greater than 5 L a day and urine volume greater than 3 L can lead to a false-positive test.16 Depression and alcohol intake may also influence UFC resulting in so-called pseudo-Cushing’s.17 A cortisol level three times above normal is suggestive of cortisol excess but would have to be confirmed with another test. When liquid chromatography–tandem mass spectrometry is used, sensitivity of UFC was 97% and specificity was 91%.
Overnight 1 mg dexamethasone suppression test (DST) involves administration of 1 mg dexamethasone between 11 pm and midnight followed by an 8 am serum cortisol level.18 The test is considered negative if the AM cortisol level is less than 1.8 μg/dL. This is a stringent criterion and may have greater sensitivity and lower specificity. It is the test of choice when an adrenal nodule/mass is an incidental finding on abdominal imaging.
Late night salivary cortisol level (LNSC) is an alternate screening test. It takes advantage of the fact that patients with true excess endogenous production of cortisol lose diurnal variability in cortisol production. On the other hand, those with pseudo-Cushing’s maintain that variability. A pooled analysis of seven studies suggested sensitivity of 93% and specificity of 96% of LNSC.19 These numbers may be lower in mild hypercortisolism and in pregnancy.
Once hypercortisolism is established, a reliable two-site IRMA (immunoradiometric) assay for ACTH is used to determine the reason for high cortisol levels. If the 8 am ACTH level is greater than 15 μg/dL, cortisol secretion is considered ACTH dependent. On the other hand, with ACTH less than 5 μg/dL, cortisol secretion is thought to be ACTH independent. While levels between 5 and 15 μg/dL are deemed indeterminate, there are little data to support these cutoffs, and the recommendations come from clinical guidelines. In ACTH-independent Cushing’s syndrome, the next step is to obtain CT or MRI imaging of the adrenal glands.
In ACTH-dependent hypercortisolism, the source of ACTH is most often the pituitary gland. The source can also be “ectopic” such as lung cancers, pancreatic tumors, etc. Pituitary adenomas secreting ACTH are somewhat resistant to negative feedback regulation by glucocorticoids while most nonpituitary ACTH-secreting malignant tumors are highly resistant to feedback suppression. Therefore, dynamic testing employing high-dose steroids is used to distinguish between ectopic and pituitary source of ACTH.
The high-dose dexamethasone suppression test can be performed in two ways: overnight18,20 or over 48 hours.
In the overnight test, a baseline AM serum cortisol level is determined and 8 mg of dexamethasone is administered at 11:00 pm followed by an 8:00 am plasma ACTH and serum cortisol measurement. A cortisol level less than 5 μg/dL is considered significant suppression. Alternatively, if the cortisol post dexamethasone is suppressed by 50% or more compared to the baseline value, the source of ACTH is thought to be pituitary.
In the 2-day high-dose dexamethasone suppression test, baseline UFC from a 24-hour urine collection is obtained. Following this, 2 mg of dexamethasone is administered every 6 hours for 48 hours. At the end of the study, urine free cortisol (24 hours) or serum cortisol is measured. Low urinary cortisol and suppressed serum cortisol UFC less than 5 μg/dL would suggest pituitary ACTH production.
The gold standard for determining source of ACTH production is inferior petrosal sinus sampling (IPSS). This is an invasive procedure in which ACTH level in inferior petrosal sinus and peripheral vein is determined simultaneously. This study can also be done following stimulation with CRH. An inferior petrosal sinus to peripheral ACTH ratio of 2:1 or greater without CRH and 3:1 or greater with use of CRH is suggestive that ACTH hypersecretion is from the pituitary.21 Laterality (left vs. right side of pituitary) can also be determined with IPSS. Imaging with MRI can then be used for localization of pituitary ACTH-secreting tumors. If it is ectopic, CT, MRI, or scintigraphy may be required for localization. Figure 21.12 summarizes the diagnostic testing algorithm.
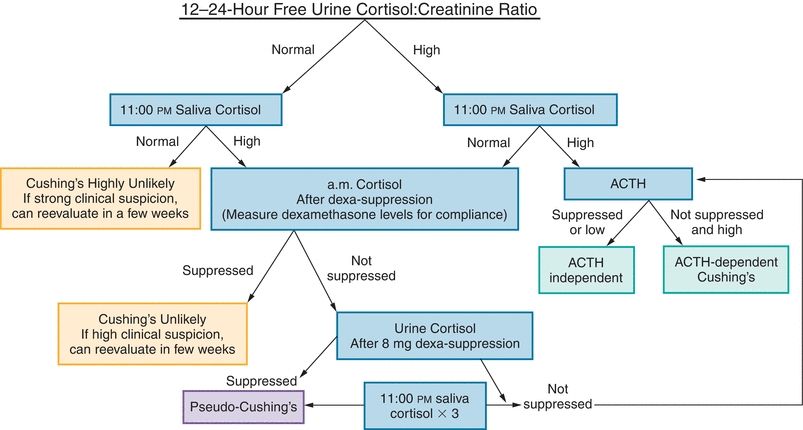
FIGURE 21.12 Cushing’s syndrome workup. Note that dexamethasone serum levels (for standard test, 2 ng/mL; suppression test, 6.5 ng/mL) can be drawn at 8:00 am (or 6 hours after the last dose) to help clarify or determine compliance. Normal dexamethasone saliva levels have not been determined. ACTH, adrenocorticotropic hormone.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


