Acute Leukemias
KEY CONCEPTS
![]() Acute leukemias are the most common malignancies in children and the leading cause of cancer-related death in patients younger than age 35 years.
Acute leukemias are the most common malignancies in children and the leading cause of cancer-related death in patients younger than age 35 years.
![]() To establish a definitive diagnosis of acute leukemia, the following diagnostic components are required: bone marrow biopsy and aspirate (with >20% blasts), cytogenetics, and immunophenotyping.
To establish a definitive diagnosis of acute leukemia, the following diagnostic components are required: bone marrow biopsy and aspirate (with >20% blasts), cytogenetics, and immunophenotyping.
![]() Several risk factors correlate with prognosis for acute lymphoblastic leukemia (ALL). Poor prognostic factors include high white blood cell count at presentation, very young or very old age at diagnosis, delayed remission induction and presence of certain cytogenetic abnormalities (e.g., Philadelphia chromosome positive [Ph+]).
Several risk factors correlate with prognosis for acute lymphoblastic leukemia (ALL). Poor prognostic factors include high white blood cell count at presentation, very young or very old age at diagnosis, delayed remission induction and presence of certain cytogenetic abnormalities (e.g., Philadelphia chromosome positive [Ph+]).
![]() For children with ALL, remission induction therapy includes vincristine, a corticosteroid, and asparaginase, with or without an anthracycline. For adults with ALL, vincristine, prednisone, and an anthracycline are given, and asparaginase is sometimes added.
For children with ALL, remission induction therapy includes vincristine, a corticosteroid, and asparaginase, with or without an anthracycline. For adults with ALL, vincristine, prednisone, and an anthracycline are given, and asparaginase is sometimes added.
![]() All patients with ALL require prophylactic therapy to prevent CNS disease because of the high risk of central nervous system relapse. The choice for therapy includes a combination of the following: cranial irradiation, intrathecal chemotherapy, or high-dose systemic chemotherapy with drugs that cross the blood–brain barrier.
All patients with ALL require prophylactic therapy to prevent CNS disease because of the high risk of central nervous system relapse. The choice for therapy includes a combination of the following: cranial irradiation, intrathecal chemotherapy, or high-dose systemic chemotherapy with drugs that cross the blood–brain barrier.
![]() Long-term maintenance therapy for 2 to 3 years is essential to eradicate residual leukemia cells and prolong the duration of remission. Maintenance therapy consists of oral methotrexate and mercaptopurine, with or without monthly pulses of vincristine and a corticosteroid.
Long-term maintenance therapy for 2 to 3 years is essential to eradicate residual leukemia cells and prolong the duration of remission. Maintenance therapy consists of oral methotrexate and mercaptopurine, with or without monthly pulses of vincristine and a corticosteroid.
![]() Disease-free survival is lower in adults with ALL and has been attributed to greater drug resistance, poor side effect tolerance with subsequent nonadherence, and possibly less-effective therapy. This population is also more likely to have Ph+ ALL, which is associated with a worse outcome, but the use of tyrosine kinase inhibitors has improved treatment results.
Disease-free survival is lower in adults with ALL and has been attributed to greater drug resistance, poor side effect tolerance with subsequent nonadherence, and possibly less-effective therapy. This population is also more likely to have Ph+ ALL, which is associated with a worse outcome, but the use of tyrosine kinase inhibitors has improved treatment results.
![]() There are several poor prognostic factors for adult acute myeloid leukemia (AML): older age, organ impairment, presence of extramedullary disease, and presence of certain cytogenetic and molecular abnormalities.
There are several poor prognostic factors for adult acute myeloid leukemia (AML): older age, organ impairment, presence of extramedullary disease, and presence of certain cytogenetic and molecular abnormalities.
![]() Therapy of AML usually includes induction therapy with an anthracycline and cytarabine. Postremission therapy is required in all patients and can include either consolidation chemotherapy with or without maintenance therapy, or hematopoietic stem cell transplantation.
Therapy of AML usually includes induction therapy with an anthracycline and cytarabine. Postremission therapy is required in all patients and can include either consolidation chemotherapy with or without maintenance therapy, or hematopoietic stem cell transplantation.
![]() It is estimated that up to 108 to 109 malignant cells remain following attainment of a complete remission. Postremission therapy with either chemotherapy or hematopoietic stem cell transplantation is essential in AML.
It is estimated that up to 108 to 109 malignant cells remain following attainment of a complete remission. Postremission therapy with either chemotherapy or hematopoietic stem cell transplantation is essential in AML.
![]() Treatment of acute promyelocytic leukemia consists of induction therapy, followed by consolidation and maintenance therapy. Induction includes tretinoin and an anthracycline; consolidation therapy consists of two to three cycles of anthracycline-based therapy; maintenance consists of pulse doses of tretinoin, mercaptopurine, and methotrexate for 2 years.
Treatment of acute promyelocytic leukemia consists of induction therapy, followed by consolidation and maintenance therapy. Induction includes tretinoin and an anthracycline; consolidation therapy consists of two to three cycles of anthracycline-based therapy; maintenance consists of pulse doses of tretinoin, mercaptopurine, and methotrexate for 2 years.
![]() Hematopoietic growth factors can be safely and effectively used with myelosuppressive chemotherapy for acute leukemias. The benefits may include reduced incidence of serious infections, reduced hospital stays, and fewer treatment delays, but do not include prolonged disease-free survival or overall survival.
Hematopoietic growth factors can be safely and effectively used with myelosuppressive chemotherapy for acute leukemias. The benefits may include reduced incidence of serious infections, reduced hospital stays, and fewer treatment delays, but do not include prolonged disease-free survival or overall survival.
The leukemias are heterogeneous hematologic malignancies characterized by unregulated proliferation of the blood-forming cells in the bone marrow. These immature proliferating leukemia cells (blasts) physically “crowd out” or inhibit normal cellular maturation in bone marrow, resulting in anemia, neutropenia, and thrombocytopenia. Leukemic blasts may also infiltrate a variety of tissues such as lymph nodes, skin, liver, spleen, kidney, testes, and the central nervous system.
Historically, leukemia has been classified based on the cell of origin and cell line maturation, and as acute or chronic based on differences in clinical presentation, rapidity of progression of the untreated disease, and response to therapy. The four major leukemias are acute lymphoblastic (or lymphocytic) leukemia (ALL), acute myeloid leukemia (AML), chronic lymphocytic leukemia, and chronic myeloid leukemia. Undifferentiated immature cells that proliferate autonomously characterize acute leukemias. Chronic leukemias also proliferate autonomously, but the cells are more differentiated and mature. Untreated, acute leukemia is fatal within weeks to months.
EPIDEMIOLOGY
It is estimated that 20,660 new cases of acute leukemia—14,590 cases of AML and 6,070 cases of ALL—will be diagnosed in the United States in 2013, accounting for 1.2% of the total cancer incidence.1 The incidence has been relatively stable for two decades. An estimated 11,800 deaths per year, representing about 2% of all cancer deaths, are caused by acute leukemias.1 ![]() Leukemia is the leading cause of cancer-related deaths in persons younger than age 20 years, but an uncommon cause of cancer-related death after age 40 years.1 Among adults, acute and chronic leukemias occur at equal rates. More than 90% of the cases of acute and chronic leukemia occur in adults. AML accounts for most cases of acute leukemia in adults, and occurs with increasing frequency in elderly patients. There are about 3.6 cases of AML and 1.4 cases of ALL per 100,000 individuals.2 The median age at diagnosis of patients with AML is about 67 years, whereas the peak age for ALL patients is 2 to 3 years.1,2 The incidence of AML increases with age from 1.8 per 100,000 in individuals younger than age 65 years to 16 per 100,000 in those 65 years or older.2,3 Acute leukemia is about 30% more common in males than in females. In the United States, acute leukemia is more common among whites than among African Americans, American Indians, and Hispanic ethnicities.2
Leukemia is the leading cause of cancer-related deaths in persons younger than age 20 years, but an uncommon cause of cancer-related death after age 40 years.1 Among adults, acute and chronic leukemias occur at equal rates. More than 90% of the cases of acute and chronic leukemia occur in adults. AML accounts for most cases of acute leukemia in adults, and occurs with increasing frequency in elderly patients. There are about 3.6 cases of AML and 1.4 cases of ALL per 100,000 individuals.2 The median age at diagnosis of patients with AML is about 67 years, whereas the peak age for ALL patients is 2 to 3 years.1,2 The incidence of AML increases with age from 1.8 per 100,000 in individuals younger than age 65 years to 16 per 100,000 in those 65 years or older.2,3 Acute leukemia is about 30% more common in males than in females. In the United States, acute leukemia is more common among whites than among African Americans, American Indians, and Hispanic ethnicities.2
Despite the low incidence, the acute leukemias are the most common malignancy in persons younger than 20 years of age, accounting for 26% of all childhood malignancies.2 About 80% of children with leukemia have ALL and 15% AML.2 Childhood ALL is about 30% more common in males than in females, peaks at 2 to 4 years of age, and is almost twice as likely to affect white children as African American children.2 The incidence of childhood AML is highest in the Hispanic population and occurs throughout childhood without any peak age period. Acute leukemia during the first year of life (infant leukemia) slightly favors ALL over AML.2,4
Chemotherapy has dramatically improved the outlook of patients with acute leukemia. More than 85% of children and young adults with acute leukemia achieve an initial complete remission (CR) of their disease. Overall, 65% to 85% of adults achieve an initial CR.3 For persons younger than 19 years of age, the 5-year survival rate is nearly 90% for ALL and 70% for AML.2,5 The prognosis of adult acute leukemia is generally worse than that of childhood leukemia, with only 30% to 40% of patients becoming long-term survivors. When all ages are included, the 5-year relative survival rate in 2008 for ALL was 66% and 25% for AML.2
ETIOLOGY
The exact cause of the acute leukemias is unknown. A multifactorial process involving genetics, environmental and socioeconomic factors, toxins, immunologic status, and viral exposures is likely. Table 111-1 summarizes the major factors that have been linked to acute leukemias. Infectious and genetic factors have the strongest associations to date.6 In pediatric ALL, a number of environmental factors are inconsistently linked to the disease: exposure to ionizing radiation, toxic chemicals, herbicides and pesticides; maternal use of contraceptives, diethylstilbestrol, or cigarettes; parental exposure to drugs (amphetamines, diet pills, and mind-altering medications), diagnostic radiographs, alcohol consumption, or chemicals before and during pregnancy; and chemical contamination of groundwater.7,8 A growing body of evidence indicates that high birthweight is a risk for ALL.6,9 Ionizing radiation and benzene exposure are the only environmental risk factors strongly associated with ALL or AML.8 A few studies have reported a possible link between electromagnetic fields of high-voltage power lines and the development of leukemia, but larger studies could not confirm this association. In most patients who develop leukemia, a cause cannot be identified.
TABLE 111-1 Risk Factors for Acute Leukemia
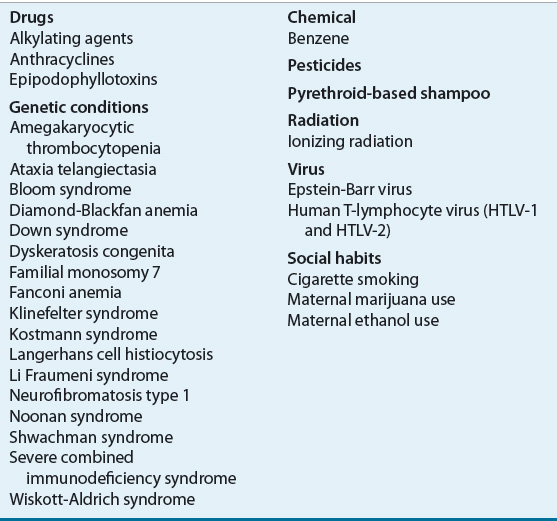
Childhood AML is associated with Hispanic ethnicity, prior exposure to alkylating agents or epipodophyllotoxins, and in utero exposure to ionizing radiation.8 Maternal alcohol consumption, parental and child organophosphate pesticide exposure, and parental benzene exposure are also associated with childhood AML. AML has been associated with both low and high birth weight.9 Adult AML has been associated with prior anthracycline exposure in addition to prior exposure to alkylating agents or epipodophyllotoxins.
PATHOPHYSIOLOGY
A basic understanding of normal hematopoiesis is needed before one can understand the pathogenesis of leukemia. eChapter 20 has a detailed discussion of hematopoiesis. Normal hematopoiesis consists of multiple well-orchestrated steps of cellular development. A pool of pluripotent stem cells undergoes differentiation, proliferation, and maturation, to form the mature blood cells seen in the peripheral circulation. These pluripotent stem cells initially differentiate to form two distinct stem cell pools. The myeloid stem cell gives rise to six types of blood cells (erythrocytes, platelets, monocytes, basophils, neutrophils, and eosinophils). Lymphoid stem cells differentiate to form natural killer cells, B lymphocytes, and T lymphocytes. Leukemia may develop at any stage and within any cell line.
Two features are common to both AML and ALL: First, both arise from a single leukemic cell that expands and acquires additional mutations, culminating in a monoclonal population of leukemia cells. Second, there is a failure to maintain a relative balance between proliferation and differentiation, so that the cells do not differentiate past a particular stage of hematopoiesis. Cells (lymphoblasts or myeloblasts) then proliferate uncontrollably. Proliferation, differentiation, and apoptosis are under genetic control, and leukemia can occur when the balance between these processes is altered.
AML probably arises from a defect in the pluripotent stem cell or a more committed myeloid precursor, resulting in partial differentiation and proliferation of immature precursors of the myeloid blood-forming cells. In older patients, trilineage leukemia occurs suggesting that the cell of origin is probably a stem or very early progenitor cell. In younger patients, a more differentiated progenitor becomes malignant, allowing maturation of some granulocytic and erythroid populations. These two forms of AML exhibit different patterns of resistance to chemotherapy, with resistance more evident in the older adults with AML. ALL is a disease characterized by proliferation of immature lymphoblasts. In this type of acute leukemia, the defect is probably at the level of the lymphopoietic stem cell or a very early lymphoid precursor.
Leukemic cells have growth and/or survival advantages over normal cells, leading to a “crowding out” phenomenon in the bone marrow. This growth advantage is not caused by more rapid proliferation as compared with normal cells. Some studies suggest that it is caused by factors produced by leukemic cells that either inhibit normal cellular proliferation and differentiation, or reduce apoptosis as compared with normal blood cells.
The types of genetic alterations that lead to leukemia have only recently become evident. The genetic defects may include (a) activation of a normally suppressed gene (protooncogene) to create an oncogene that produces a protein product that signals increased proliferation; (b) loss of signals for the blood cell to differentiate; (c) loss of tumor suppressor genes that control normal proliferation; and (d) loss of signals for apoptosis. Most normal cells are programmed to die eventually through apoptosis, but the appropriate programmed signal is often interrupted in cancer cells, leading to continued survival, replication, and drug resistance. Signal transduction, RNA transcription, cell-cycle control factors, cell differentiation, and programmed cell death may all be affected.
LEUKEMIA CLASSIFICATION
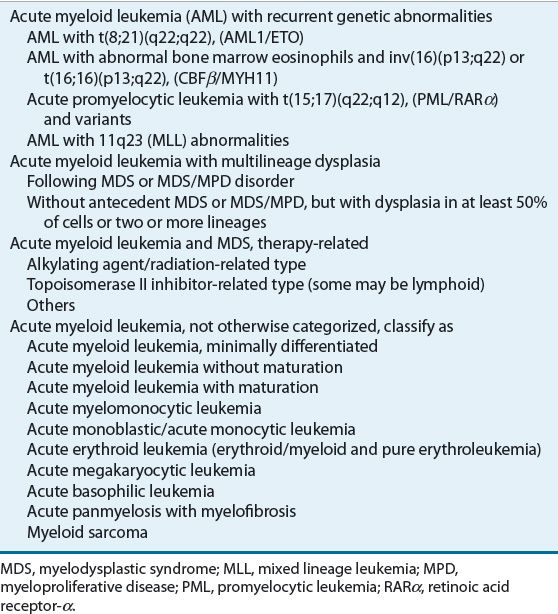
In 2001, the World Health Organization (WHO), in collaboration with the Society for Hematopathology and the European Association of Haematopathology, proposed a new classification system for myeloid neoplasms. This new classification system incorporates not only morphologic findings, but also genetic, immunophenotypic, cytochemical, and clinical features. About 40% to 50% of adult patients with AML have no detectable chromosomal abnormality on standard cytogenetic analysis, but the percent increases with age.10 The WHO classification attempts to formally incorporate the relationship between AML and myelodysplastic syndrome (MDS). In 2008, this classification was revised (Table 111-2) and is being used routinely for children and adults.11 The WHO classification defines acute leukemias as more than 19% blasts in the marrow or blood.
TABLE 111-2 World Health Organization Classification of Acute Myeloid Leukemia
Lymphoblast analysis is used to classify ALL. Immunophenotype is determined by flow cytometry that analyzes specific antigens, known as clusters of differentiation (often abbreviated “CD”), present on the surface of hematopoietic cells. Although no leukemia-specific antigens have been identified, the pattern of cell-surface antigen expression reliably distinguishes between lymphoid and myeloid leukemia. The immunophenotype defines the cell of origin. The major phenotypes are mature B-cell, precursor B-cell, and T-cell disease; however, the WHO classifies ALL as either B lymphoblastic or T lymphoblastic. About 80% of childhood ALL derives from precursor B cells and about 15% from T cells; the remainder is either mixed lineage or from mature B cells. T-cell ALL is more common in teenage males.
Leukemias may also be described by cytogenetic abnormalities. Chromosome alterations include numerical (hyperdiploidy and hypodiploidy), and structural abnormalities due to exchanges of genetic information within (inversion) or between (translocation) chromosomes. Unique translocations can identify specific subtypes of acute leukemia. Twenty-five percent of children with precursor B-cell ALL have the ETV6-RUNX1 (formerly TEL-AML1) fusion gene generated by the t(12;21) (p13;q22) chromosomal translocation.6,12 This translocation appears to endow the preleukemia cell with altered self-renewal and survival properties. The most common translocation in adult ALL, occurring in 25% of patients, is the t(9;22) or Philadelphia chromosome positive (Ph+), which causes fusion of the BCR signaling protein to the ABL nonreceptor tyrosine kinase, resulting in constitutive tyrosine kinase activity. More than 50% of childhood T-cell ALL have activating mutations of the NOTCH1 gene that encodes for a transmembrane receptor implicated in regulation of T cell development.6,12 Acute promyelocytic leukemia (APL) is characterized by a specific translocation between chromosomes 15 and 17: t(15;17). Molecular tests may be used to identify products of specific translocations, such as promyelocytic leukemia (PML) retinoic acid receptor-α (RARα) in APL and AML1-ETO and CBFβ/MYH 11 in other subtypes of AML.
A number of factors may affect the cytogenetics of AML in adults. First, in about 5% of patients, simultaneous blood and marrow samples demonstrate normal cytogenetics versus abnormal cytogenetics, respectively.10 Second, central cytogenetic analysis is done in multicenter trials because of variability in specimen examination. A small number of patients may have a normal karyotype on standard review, but carry fusion genes, which are identical to those of translocations or inversions. These insertions of very small chromosome segments do not alter chromosome morphology but may affect outcome.
CLINICAL PRESENTATION AND DIAGNOSIS
Common signs and symptoms at presentation result from malignant cells that replace and suppress normal hematopoietic progenitor cells and infiltrate into extramedullary spaces. Many of the signs and symptoms result from low blood cells. Thrombocytopenia can result in bruising, petechiae, and bleeding; low red blood cells can result in fatigue and loss of energy; and low white blood cells can result in signs and symptoms of infection such as fever, chills, and rigors. Patients with ALL may rarely present with small blue-green collections of leukemia cells under the skin called chloromas.
![]() In addition to clinical presentation, laboratory and pathology evaluations are required for a definitive diagnosis of leukemia. An abnormal complete blood count is usually the diagnostic test that initiates a leukemia workup. The most important test is a bone marrow biopsy and aspirate, which is submitted to hematopathology for numerous evaluations. A lumbar puncture is performed to determine if there are blasts in the central nervous system (CNS). A chest radiograph is performed to screen for a mediastinal mass (most common in T-cell disease).
In addition to clinical presentation, laboratory and pathology evaluations are required for a definitive diagnosis of leukemia. An abnormal complete blood count is usually the diagnostic test that initiates a leukemia workup. The most important test is a bone marrow biopsy and aspirate, which is submitted to hematopathology for numerous evaluations. A lumbar puncture is performed to determine if there are blasts in the central nervous system (CNS). A chest radiograph is performed to screen for a mediastinal mass (most common in T-cell disease).
CLINICAL PRESENTATION
ACUTE LYMPHOBLASTIC LEUKEMIA
Risk Classification
Many clinical and biologic features at diagnosis are associated with response to treatment, as measured by the CR rate, duration of remission, and long-term survival. The patient’s response to initial therapy is also strongly associated with response to treatment. Identification of these risk factors allows the clinician to better understand the disease and to tailor treatment according to risk of disease recurrence (i.e., risk-adapted therapy). For example, if a patient has many clinical and laboratory features that are associated with a good response to chemotherapy (“standard risk”), then the clinician may choose to give less-intensive therapy to reduce the risk of long-term adverse effects. Conversely, if a patient is unlikely to respond well to standard therapy (high-risk or very-high-risk disease), then the clinician may choose to give more intensive chemotherapy. The factors can be grouped as follows: patient characteristics at diagnosis, leukemic cell features at diagnosis, and patient response to initial therapy.
Patient Characteristics
![]() The National Cancer Institute (NCI) developed an ALL risk stratification to create a standard for comparison in children.13 Induction therapy is initially selected based on this classification, which divides children into standard- or high-risk categories based on age and initial white blood cell (WBC) count (Table 111-3a). Age remains an independent predictor of outcome with children aged 1 to 9 years having the best event-free survival (EFS). This is mostly explained by the more frequent occurrence of the ETV6-RUNX1 translocation and hyperdiploidy (>51 chromosomes) in addition to increased drug sensitivity in this age group.14 Presence of CNS disease at diagnosis is associated with a higher relapse rate. About 2% of males have testicular disease at diagnosis, but cooperative groups vary in whether this is an adverse prognostic factor. Patients with Down syndrome tend to have lower EFS, but this is mostly attributed to higher treatment-related morbidity and mortality.
The National Cancer Institute (NCI) developed an ALL risk stratification to create a standard for comparison in children.13 Induction therapy is initially selected based on this classification, which divides children into standard- or high-risk categories based on age and initial white blood cell (WBC) count (Table 111-3a). Age remains an independent predictor of outcome with children aged 1 to 9 years having the best event-free survival (EFS). This is mostly explained by the more frequent occurrence of the ETV6-RUNX1 translocation and hyperdiploidy (>51 chromosomes) in addition to increased drug sensitivity in this age group.14 Presence of CNS disease at diagnosis is associated with a higher relapse rate. About 2% of males have testicular disease at diagnosis, but cooperative groups vary in whether this is an adverse prognostic factor. Patients with Down syndrome tend to have lower EFS, but this is mostly attributed to higher treatment-related morbidity and mortality.
TABLE 111-3a National Cancer Institute (NCI) Risk Classification (Smith 1996)

Race is controversial, with older studies indicating worse outcomes for minorities. Male race and obesity have been associated with worse outcome in cooperative group studies, but not single-institution studies.6 Hepatosplenomegaly and mediastinal mass are both associated with worse outcomes.
Leukemic Cell Characteristics
With current therapy, the cell of origin no longer has a prognostic significance as therapy is tailored to account for this historical difference. Several chromosomal (cytogenetic) abnormalities are associated with prognosis. Children with ALL have an average of six DNA copy number alterations.12 Favorable outcomes are associated with three copies of chromosomes 4 and 10, high hyperdiploidy (51-65 chromosomes), and the ETV6-RUNX1 cryptic translocation, t(12;21).12 NOTCH1 and FBXW7 mutations confer a favorable prognosis for patients with T-cell disease.12 The Philadelphia chromosome is present in 3% to 5% of children and 25% of adults and is historically associated with a poor prognosis.15 The mixed lineage leukemia (MLL) gene rearrangement (11q23), intrachromosomal amplification of chromosome 21 (iAMLP21), and hypodiploidy (<44 chromosomes) are associated with a poorer prognosis.16
Initial Response to Therapy
The strongest prognostic factor for outcome for ALL is response to therapy. Both the rapidity of response and the level of residual disease at the end of induction therapy are associated with long-term outcome. Children with a reduction of bone marrow lymphoblasts within 14 days of initiating chemotherapy (rapid early responders) have a more favorable prognosis. Molecular measurement of sub-clinical minimal residual disease (MRD) by either flow cytometry or polymerase chain reaction has enabled detection of leukemic cells not visible on morphologic examination to assess treatment response and detect relapse in children and adults.17 This technique allows detection of 1 leukemia cell in 10,000 normal cells, which is about 100-fold more sensitive than morphologic examination.17 If MRD is detected at the end of induction therapy, the clinician may decide to give more intensive therapy to decrease the risk of relapse.
The Children’s Oncology Group uses a risk- and response-based classification of childhood ALL (Fig. 111-1).18 This classification system uses the NCI risk assignment to initially categorize patients into standard- or high-risk groups (Table 111-3a). Following induction therapy, risk is reclassified based on the rapidity and completeness of response to therapy, the presence or absence of cytogenetic abnormalities or CNS involvement (Table 111-3b). Patients are then reclassified as low risk, standard risk, high risk, or very-high risk (Fig. 111-1). Patients who are initially high risk do not have therapy reduced, but may have it intensified to very-high risk as discussed below.
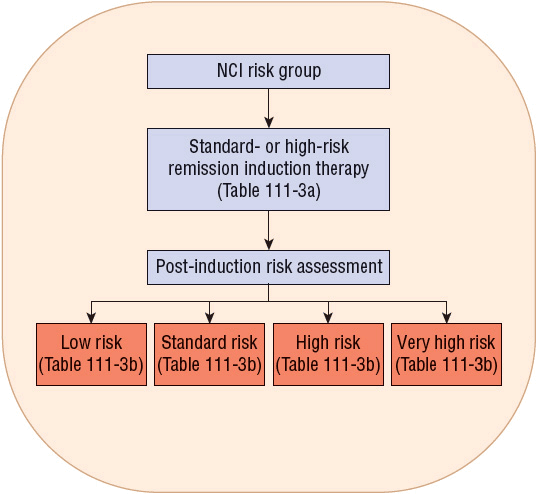
FIGURE 111-1 Pediatric precursor B-cell acute lymphoblastic leukemia risk classification. (NCI, National Cancer Institute.)
TABLE 111-3b Pediatric Precursor B-Cell Acute Lymphoblastic Leukemia Risk Classification
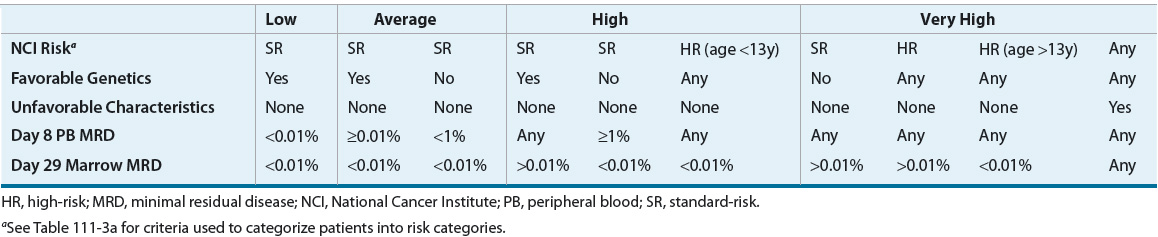
Children are classified as low risk and will have therapy reduced if they have trisomy 4 and 10 or the ETV6-RUNX1 cryptic translocation with less than 0.01% MRD on day 8 peripheral blood and day 29 bone marrow samples. Children with testicular disease, >5% blasts in the bone marrow by day 15, MRD ≥0.01% at day 29, or who received steroids prior to diagnosis have postinduction therapy intensified and are classified as high risk. Childhood precursor B-ALL with more than five WBCs and blasts present in the cerebrospinal fluid (CSF), Ph+ disease, hypodiploidy, iAMLP21, induction failure, or MLL gene rearrangement have therapy intensified and are considered very-high risk. Infant ALL, trisomy 21, or childhood T-cell ALL have unique risk classification schemas. Children with T-cell leukemia historically have an inferior response to standard-risk therapy and are automatically categorized as high risk to receive augmented therapy and T cell targeted therapy. T-cell and mature B-cell disease are favorable phenotypes in adults.6 Age is inversely associated with prognosis in patients with Ph+ ALL.6
TREATMENT
Acute Lymphoblastic Leukemia
Desired Outcomes
The short-term goal for ALL treatment is to rapidly achieve a complete clinical and hematologic remission. A CR is defined as the disappearance of all physical and bone marrow evidence (normal cellularity with <5% blasts) of leukemia, with restoration of normal hematopoiesis. After a CR is achieved, the goal is to maintain the patient in continuous CR. In general, a child is considered to be “cured” after being in continuous CR for 5 to 10 years.
Successful treatment of ALL was first developed in children. Current regimens induce clinical remission in 96% to 99% of children with ALL.6 MRD is a strong predictor of relapse in ALL. Children with MRD in the bone marrow at the end of induction have a 5-year EFS of 59% versus 88% in children without.19 Children with low-risk disease have a 5-year EFS of more than 95%.21 The 5-year EFS for average-risk disease is 90 to 95%.21 The 5-year EFS is nearly 90% for high-risk childhood B-precursor and T-cell ALL including rapid and slow responders. Children with very-high-risk disease have a 5-year EFS of less than 80%.21 Response to treatment is determined by intrinsic drug sensitivity and the patient’s pharmacogenomics and pharmacodynamics, treatment received, and treatment adherence. Cure rates in children have risen from less than 5% with treatments used in the 1960s to about 90% by 2005.22 The reason for this improvement lies largely in improved scheduling of existing drugs, as relatively few new drugs have come to the market since the 1960s.
Although treatment results with adult ALL are worse than those with childhood ALL, recent use of aggressive chemotherapy in adult ALL has increased the CR rate from 60% to 85%. Long-term EFS in this population, however, remains low (between 30% and 40%) because a higher proportion of adults present with poor-risk disease. CR rates and EFS vary according to a number of poor prognostic factors and certain types of ALL are associated with a very poor outcome.
Treatment Phases
Therapy for childhood ALL is divided into five phases: (a) induction, (b) consolidation therapy, (c) interim maintenance, (d) delayed intensification, and (e) maintenance therapy (Fig. 111-2). CNS prophylaxis is a mandatory component of ALL treatment regimens and is administered longitudinally during all phases of treatment. The total duration of treatment is 2 to 3 years.
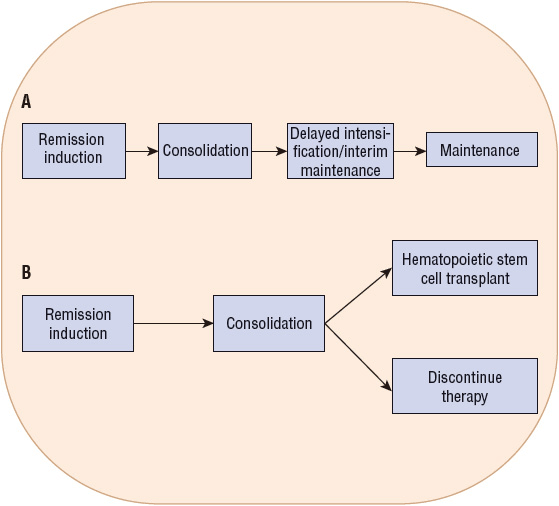
FIGURE 111-2 Treatment algorithm for (A) acute lymphoblastic leukemia and (B) acute myeloid leukemia.
Induction
The goal of induction is to rapidly induce a complete clinical and hematologic remission. ![]() The CR rate is 98% for standard-risk children treated with vincristine, a glucocorticoid (dexamethasone or prednisone), and asparaginase or pegaspargase.22 Most treatment protocols add daunorubicin to induction (four-drug induction) for high-risk or very-high-risk ALL, while others add cyclophosphamide, methotrexate, or cytarabine. Most children achieve a CR in 4 weeks, which classifies them as rapid early responders. Those who have a M2 (5% to 25% blasts) or M3 (>25% blasts) marrow on day 15 of induction or have positive MRD at day 29 are classified as slow early responders and receive intensified therapy. Only 2% to 3% of children fail induction therapy, but the therapy results in a 10-year survival rate of 32%.23
The CR rate is 98% for standard-risk children treated with vincristine, a glucocorticoid (dexamethasone or prednisone), and asparaginase or pegaspargase.22 Most treatment protocols add daunorubicin to induction (four-drug induction) for high-risk or very-high-risk ALL, while others add cyclophosphamide, methotrexate, or cytarabine. Most children achieve a CR in 4 weeks, which classifies them as rapid early responders. Those who have a M2 (5% to 25% blasts) or M3 (>25% blasts) marrow on day 15 of induction or have positive MRD at day 29 are classified as slow early responders and receive intensified therapy. Only 2% to 3% of children fail induction therapy, but the therapy results in a 10-year survival rate of 32%.23
Prednisone has historically been the primary glucocorticoid used in pediatric ALL regimens.24 Dexamethasone is now being used in most standard-risk protocols because of its longer duration of action and higher CSF penetration compared to prednisone.6,22,24 When dexamethasone is used in place of prednisone, absolute EFS improves by 5% to 9% and the risk of CNS relapse decreases by 2% to 4%.14,24,25 However, dexamethasone increases the risk of side effects such as osteonecrosis, mood alteration, steroid myopathy, hyperglycemia, and infections.24–26 Patients older than 10 years of age are particularly prone to osteonecrosis and receive prednisone instead of dexamethasone to minimize this side effect. Low serum albumin prolongs dexamethasone exposure and may contribute to increased toxicity.14 Since patients with Down’s syndrome have increased infections and mortality with dexamethasone, these patients receive prednisone.
Clinical Controversy…
The incidence of transient hyperglycemia during pediatric ALL induction therapy has increased. In a recent study, 20% of children had transient hyperglycemia, defined as at least two random serum glucose levels greater than or equal to 200 mg/dL (11.1 mmol/L).27 The incidence was higher (42%) in children older than 10 years of age. The risk was higher for patients with a body mass index (BMI) greater than or equal to the 95th percentile and those receiving asparaginase as compared to pegaspargase.
Asparaginase has historically been available in three forms. Asparaginase (no longer manufactured in the United States) and pegaspargase are isolated from Escherichia coli while Erwinia asparaginase is isolated from Erwinia chrysanthemi. A recombinant E. coli asparaginase and a pegylated form of recombinant Erwinia asparaginase are currently in clinical trials. Pegaspargase is pegylated E. coli asparaginase; pegylation prolongs its duration of activity and allows it to be given less frequently. Pegaspargase is used in most protocols and is preferred over asparaginase because of fewer intramuscular injections, decreased antibody formation, and superior response rates. Pegaspargase is also approved for IV administration. The use of prolonged intensive asparaginase treatment compared with shorter treatment increases absolute EFS by 4% to 17%.28
Asparaginase products are the chemotherapeutic agents most likely to cause hypersensitivity reactions. Depending on the product and coadministered steroid, 8% to 42% of patients develop antibodies to asparaginase. Reactions usually occur during postinduction phases of therapy when asparaginase has not been given for a prolonged period of time.28 The reaction is delayed with pegaspargase and usually occurs 6 to 12 hours following a dose. The hypersensitivity reaction to pegaspargase may also be prolonged and frequently requires hospitalization for 5 to 7 days. Erwinia asparaginase is currently only used for patients who are allergic to other available forms. Patients may also have “silent hypersensitivity” in which they develop neutralizing antibodies that can rapidly inactivate the asparaginase, but the patient does not have a clinical hypersensitivity reaction. Therefore, many centers prefer switching from E. coli derived products to Erwinia asparaginase to avoid this scenario.
Central Nervous System Prophylaxis
CNS prophylaxis is incorporated throughout all phases of therapy. The rationale for CNS prophylaxis is based on two observations. First, many chemotherapeutic agents do not readily cross the blood–brain barrier. Second, results from early clinical trials of ALL showed that 50% to 85% of patients with ALL and no CNS involvement at diagnosis experienced a CNS relapse.7 These observations indicate that the CNS is a potential sanctuary for leukemic cells and undetectable leukemic cells are present in the CNS in many patients at the time of diagnosis. Detectable CNS involvement at the time of diagnosis occurs in 3% of children with ALL.14 Factors that are associated with an increased risk of CNS involvement at diagnosis in children include a high initial WBC count, T-cell phenotype, mature B-cell phenotype, age ≤1 year, African American race, thrombocytopenia, lymphadenopathy, and hepatomegaly or splenomegaly.22
The goal of CNS prophylaxis is to eradicate undetectable leukemic cells from the CNS while minimizing neurotoxicity and late effects. Leukemic meningitis is more easily prevented than treated. Once CNS relapse has occurred, patients are at increased risk of bone marrow relapse and death from refractory leukemia. Initial trials of childhood ALL in the 1960s established craniospinal irradiation as the standard for prevention of CNS relapse.22 However, this approach is associated with long-term sequelae including neuropsychological deficits, precocious puberty, osteoporosis, decreased intellect, thyroid dysfunction, brain tumors, short stature, and obesity. Subsequent trials have demonstrated that irradiation may be replaced by frequent administration of intrathecal chemotherapy in children with ALL.22 Some centers may treat children with CNS disease at diagnosis or very-high-risk disease with cranial radiation.
![]() The CNS prophylaxis regimen is selected based on efficacy, toxicity, and risk of CNS disease. Intrathecal chemotherapy, cranial irradiation, dexamethasone, and high-dose IV methotrexate or cytarabine can be used to treat or prevent CNS disease. Current treatment approaches have reduced isolated CNS relapses to less than 5% among children.29 Risk factors for CNS relapse include male sex, hepatomegaly, T-cell phenotype, CNS2 disease (the presence of leukemic blasts in a CSF sample that contains <5 WBC/mm3), age younger than 2 years or older than 6 years, and a bloody diagnostic lumbar puncture.6,29 Intrathecal therapy consists of methotrexate and cytarabine, given either alone or in combination. When given together, hydrocortisone is commonly added (triple intrathecal therapy) to decrease the incidence of arachnoiditis. For standard-risk ALL, triple intrathecal therapy decreased relative CNS relapse rates by 30% in comparison to intrathecal methotrexate and no effect on EFS and worse overall survival (OS).29 Triple intrathecal therapy is typically reserved for children with refractory CNS disease. The doses of intrathecal chemotherapy used for childhood ALL are age-based because of differences in the volume of CSF at various ages. For example, intrathecal methotrexate is dosed as 8 mg if <2 years, 10 mg for 2 to 2.99 years, 12 mg for 3 to 8.99 years, and 15 mg for ≥9 years. Liposomal cytarabine induces CNS remission in 57% of relapsed patients, but is associated with a high incidence of arachnoiditis and other CNS-related adverse effects.30 Currently its use is limited to refractory or relapsed CNS disease in children.
The CNS prophylaxis regimen is selected based on efficacy, toxicity, and risk of CNS disease. Intrathecal chemotherapy, cranial irradiation, dexamethasone, and high-dose IV methotrexate or cytarabine can be used to treat or prevent CNS disease. Current treatment approaches have reduced isolated CNS relapses to less than 5% among children.29 Risk factors for CNS relapse include male sex, hepatomegaly, T-cell phenotype, CNS2 disease (the presence of leukemic blasts in a CSF sample that contains <5 WBC/mm3), age younger than 2 years or older than 6 years, and a bloody diagnostic lumbar puncture.6,29 Intrathecal therapy consists of methotrexate and cytarabine, given either alone or in combination. When given together, hydrocortisone is commonly added (triple intrathecal therapy) to decrease the incidence of arachnoiditis. For standard-risk ALL, triple intrathecal therapy decreased relative CNS relapse rates by 30% in comparison to intrathecal methotrexate and no effect on EFS and worse overall survival (OS).29 Triple intrathecal therapy is typically reserved for children with refractory CNS disease. The doses of intrathecal chemotherapy used for childhood ALL are age-based because of differences in the volume of CSF at various ages. For example, intrathecal methotrexate is dosed as 8 mg if <2 years, 10 mg for 2 to 2.99 years, 12 mg for 3 to 8.99 years, and 15 mg for ≥9 years. Liposomal cytarabine induces CNS remission in 57% of relapsed patients, but is associated with a high incidence of arachnoiditis and other CNS-related adverse effects.30 Currently its use is limited to refractory or relapsed CNS disease in children.
Patients with T-cell leukemia have an increased incidence of CNS disease and usually receive systemic therapy that penetrates the CNS such as high-dose methotrexate.26 A WBC count greater than 100,000 cells/mm3 (100 × 109/L) is associated with an increased risk of CNS relapse.6 Patients with T-cell disease have lower methotrexate polyglutamate accumulation and addition of high-dose methotrexate in these patients results in fewer CNS relapses and improves EFS.14
Consolidation Therapy
Consolidation therapy in ALL is started after a CR has been achieved, and refers to continued intensive chemotherapy in an attempt to eradicate clinically undetectable disease in order to secure (consolidate) the remission. Regimens usually incorporate either non–cross-resistant drugs that are different from the induction regimen, or more dose-intensive use of the same drugs.
Randomized trials show that consolidation therapy clearly improves patient outcome in children, but its benefit in adults is less clear.22 The relative benefit of individual components of treatment regimens is difficult to demonstrate because of the overall complexity of therapy in ALL. Standard consolidation lasts 4 weeks and usually consists of vincristine, mercaptopurine, and intrathecal methotrexate. Children with testicular disease usually receive radiation during this phase of therapy if a complete clinical response in the testes is not achieved by the end of induction. In children, the intensity of consolidation therapy is based on the child’s initial risk classification and response to induction therapy. Patients who respond slowly to induction therapy are at higher risk of relapse if they are not treated on more aggressive regimens. Children who are slow early responders or have high-risk disease benefit from intensified consolidation that includes the addition of vincristine and pegaspargase to standard therapy (cyclophosphamide, low-dose cytarabine, mercaptopurine).20
Children with Ph+ ALL, infants with MLL, or children who only achieve a partial remission may undergo allogeneic hematopoietic stem cell transplantation (HSCT) in first remission if a suitable donor is available. Nelarabine is a prodrug of ara-G that preferentially accumulates in T lymphoblasts as ara-guanosine triphosphate (GTP). Children and young adults in first bone marrow relapse had a 55% complete or partial response in the phase II trial.30 Nelarabine added to a high-risk backbone for initial therapy of childhood T-cell ALL in Children’s Oncology Group (COG) AALL00P2 was well tolerated and had a 5-year EFS of 69% in slow early responders compared to 35% to 51% in historical controls.31
Reinduction (Delayed Intensification and Interim Maintenance)
One or two delayed intensification phases separated by low-intensity interim maintenance cycles can be added to maintain remission and to decrease cumulative toxicity. Delayed intensification usually consists of drugs used during induction and consolidation or agents that lack cross-resistance with those already received such as cyclophosphamide, methotrexate, and limited amounts of doxorubicin. The methotrexate dose is variable; standard-risk children generally receive 1 to 2 g/m2 while those with T-cell disease generally receive 5 g/m2. Interim maintenance usually consists of dexamethasone, vincristine, weekly methotrexate, mercaptopurine, and intrathecal methotrexate. Delayed intensification improves EFS for standard-risk children.14,20 Delayed intensification with dose intensification improved EFS and decreased late relapses for high-risk childhood ALL, but there was no additional benefit for two delayed intensification cycles.20 Children on the intensified arms of the study received significantly more antimicrobial drugs, blood products, and parenteral nutrition but had no increase in treatment-related mortality.20 The antimetabolite-based regimens may have a reduced risk of late toxicities, but the more intensive regimens appear to result in better survival for some patients, especially those with higher risk disease.
Maintenance Therapy
![]() Maintenance therapy allows long-term drug exposure to slowly dividing cells, allows the immune system time to eradicate leukemia cells, and promotes apoptosis (programmed cell death). The goal of maintenance therapy is to further eradicate residual leukemic cells and prolong remission duration. Although maintenance therapy is clearly beneficial in childhood ALL, the benefit in adults has only recently been demonstrated.
Maintenance therapy allows long-term drug exposure to slowly dividing cells, allows the immune system time to eradicate leukemia cells, and promotes apoptosis (programmed cell death). The goal of maintenance therapy is to further eradicate residual leukemic cells and prolong remission duration. Although maintenance therapy is clearly beneficial in childhood ALL, the benefit in adults has only recently been demonstrated.
Maintenance therapy usually consists of daily mercaptopurine and weekly methotrexate for 12-week courses, at doses that produce relatively little myelosuppression, with monthly “pulses” of vincristine and a steroid for 5 days per month. Based on the results of studies that show a trend toward an increase in late relapse (excluding isolated testicular relapse) among male children treated for 2 years versus 3 years, some centers treat female children for 2 years while males receive maintenance for a total of 3 years of therapy.
Interpatient variability in the pharmacokinetics of oral methotrexate and mercaptopurine may also be an important determinant of the effectiveness and toxicity of maintenance therapy. Patients who take oral methotrexate and mercaptopurine on an evening versus a morning schedule appear to have a superior outcome. Mercaptopurine cannot be given with milk or milk products because of the presence of xanthine oxidase. Children whose adherence rate taking mercaptopurine is less than 95% have a 2.5-fold higher risk of suffering a relapse.32 Factors associated with nonadherence include single-parent household, adolescence, lower socioeconomic status, and Hispanic ethnicity.32 To account for the interpatient variability, most clinicians will titrate the dose of either agent to maintain an absolute neutrophil count of 500 to 1,500 cells/mm3 (0.5 to 1.5 × 109/L).6 Some protocols overcome bioavailability and poor adherence issues by administering methotrexate IV or intramuscularly. The importance of these pharmacokinetic issues in adults is not well defined.
Philadelphia Chromosome Positive Acute Lymphoblastic Leukemia
Ph+ ALL has a 45% 7-year EFS and has historically been treated as very-high-risk disease.15 This includes a four-drug induction. The addition of continuous imatinib mesylate, a signal transduction inhibitor that inhibits BCR-ABL kinase, through all phases of treatment resulted in a 3-year EFS of 80% in comparison to 35% for historical controls.33 The results for patients receiving chemotherapy with imatinib were equivalent to those receiving HSCT. Imatinib is currently incorporated into childhood treatment trials for Ph+ ALL in Europe and the United States. Trials are ongoing with the more potent tyrosine kinase inhibitors, nilotinib and dasatinib. Imatinib has been incorporated into consolidation for children with Ph+ disease.
Acute Lymphoblastic Leukemia in Infants
ALL and AML in infants younger than 1 year of age account for less than 5% of the reported acute leukemias in childhood, but they are associated with poor outcomes. OS in infant ALL is 30% to 50%.34 About 70 to 80% of infants with acute leukemia have t(4;11) involving the MLL gene.34 MLL gene rearrangements are associated with worse outcome, with only 18% to 34% 4- to 8-year EFS.34 Infants with ALL are more likely to present with a high WBC count, hepatosplenomegaly, and CNS disease.34 Age younger than 6 months at diagnosis and poor response to prednisone alone given prior to starting other agents are poor prognostic indicators.34 Infants with MLL gene rearrangements are more likely to overexpress FLT3, a tyrosine kinase implicated in leukemogenesis. Current trials are testing the efficacy of FLT3 inhibitors in infants with MLL gene rearrangements. Patients with infant ALL may have greater drug resistance to asparaginase, vincristine, and corticosteroids, but increased sensitivity to cytarabine and cladribine.34 Although intensive regimens such as high-dose methotrexate and high-dose cytarabine have improved survival rates they resulted in unacceptably high mortality rates. Lack of pharmacokinetic data for chemotherapy in infants has contributed to toxicity from inappropriate dosing of doxorubicin and vincristine. The use of allogeneic HSCT for infants with ALL remains controversial because of a lack of donors, concerns over the long-term toxicity of total body irradiation, and excessive mortality in some series. The Interfant-99 study showed a benefit for HSCT in a subset of infants with MLL gene rearrangement, age younger than 6 months of age, or WBC greater than 300,000 cells/mm3 (300 × 109/L).35
Acute Lymphoblastic Leukemia in Adolescents and Young Adults
Although ALL is relatively uncommon in adolescents and young adults (AYA) (15 to 39 years AYA), the outcomes are generally worse than for childhood ALL. ALL in AYA has a higher frequency of T-cell immunophenotype and a lower frequency of the t(12;21) (p13;q22) cryptic translocation responsible for hyperdiploidy and the ETV6-RUNX1 fusion gene; about 5% to 7% of ALL in AYA have Ph+ disease (higher than children, but lower than older adults).12,36 A retrospective comparison of 16- to 20-year-old patients treated on pediatric versus adult protocols in the United States resulted in identical CR rates, but the 7-year EFS favored the patients treated on pediatric regimens (64% vs. 34%).36 Patients treated on the pediatric regimens also had a 10% lower CNS relapse rate. The adult regimens studied were more myelosuppressive, while the pediatric regimens intensified steroids and asparaginase and included earlier and more intensive CNS-directed therapy. The adult regimens also had a higher risk of late effects due to higher doses of daunorubicin and use of cyclophosphamide. A current adult intergroup study is using a pediatric regimen for AYA patients and will be able to evaluate some of the other potential reasons for the outcome disparity, such as adherence and psychosocial differences. AYA patients may receive treatment based on an adult or pediatric regimen depending on institutional preferences, but the trend is shifting toward pediatric regimens.37
Acute Lymphoblastic Leukemia in Adults
Treatment risk stratification for adult patients differs depending on age and Philadelphia chromosome status. The National Comprehensive Cancer Network (NCCN) guidelines recommend different strategies for adolescents and young adults (AYA) ages 15 to 39, adults 40 to 65, and adults older than 65 years with or without poor performance status.37 While complete remission is achieved in 70% to 90% of adults with a four-drug induction regimen containing daunorubicin or doxorubicin, vincristine, an asparaginase formulation, and prednisone, long-term EFS is considerably lower and achieved in only 30% to 40% of patients.38 ![]() Poorer outcomes in adults have been attributed to differences in cytogenetic abnormalities, greater drug resistance, higher risk of treatment-related adverse effects with subsequent nonadherence, and possibly less effective therapy. The value of adding more agents to the basic four-drug induction regimen or higher doses of drugs in the remission induction regimen is not clear. Several different regimens are considered appropriate to use as first-line therapies in adults including the Cancer and Leukemia Group B (CALGB) 8811 (Larson regimen), Eastern Cooperative Oncology Group (ECOG) 2993, or Linker regimen.39–41 Some studies suggest that high-dose methotrexate and cytarabine alternating with fractionated cyclophosphamide plus vincristine, doxorubicin, and dexamethasone (hyperCVAD) may improve response and survival in adults with ALL.42 A considerable number of ALL cases occur in patients older than age 65 years, and treatment of this group of patients is an even greater challenge. The response to therapy and durability of response is less than in all other populations. Treatment-related mortality rates during remission induction therapy are also higher in this population.
Poorer outcomes in adults have been attributed to differences in cytogenetic abnormalities, greater drug resistance, higher risk of treatment-related adverse effects with subsequent nonadherence, and possibly less effective therapy. The value of adding more agents to the basic four-drug induction regimen or higher doses of drugs in the remission induction regimen is not clear. Several different regimens are considered appropriate to use as first-line therapies in adults including the Cancer and Leukemia Group B (CALGB) 8811 (Larson regimen), Eastern Cooperative Oncology Group (ECOG) 2993, or Linker regimen.39–41 Some studies suggest that high-dose methotrexate and cytarabine alternating with fractionated cyclophosphamide plus vincristine, doxorubicin, and dexamethasone (hyperCVAD) may improve response and survival in adults with ALL.42 A considerable number of ALL cases occur in patients older than age 65 years, and treatment of this group of patients is an even greater challenge. The response to therapy and durability of response is less than in all other populations. Treatment-related mortality rates during remission induction therapy are also higher in this population.
While the overall incidence of Ph+ positive disease is 25% in adults, the incidence rises with increasing age to over 40% in adults older than the age of 50 years.43 Traditionally, treatment outcomes for patients with Ph+ ALL has been extremely poor with reported OS rates of less than 20% and for those continuing to allogeneic HSCT a 2-year OS of 40% to 50%. As compared with historical control patients treated with standard chemotherapy alone, the addition of BCR-ABL tyrosine kinase therapy to chemotherapy was associated with an increased CR and OS.44,45 No randomized trials have compared imatinib and conventional chemotherapy versus conventional chemotherapy alone. The CR rates seen with tyrosine kinase inhibitors appear to be more durable and allow more patients with Ph+ disease to proceed to allogeneic HSCT. This approach also appears to be tolerated in elderly patients.46,47 For patients older than 65 years of age or for those with a poor performance status, induction regimens may include concurrent chemotherapy with a tyrosine kinase inhibitor, either alone or combined with corticosteroids. Based on these data, the combination of imatinib with concurrent chemotherapy is currently considered as the standard of care for first-line therapy.
Dasatinib has also been studied in combination with concurrent chemotherapy as first-line therapy with demonstrated efficacy.48 Other newer BCR-ABL tyrosine kinase inhibitors, nilotinib, bosutinib, and ponatinib, have also been evaluated in patients with imatinib-resistant Ph+ leukemias. Responses can be achieved, but more data are needed to evaluate their specific role in the treatment of Ph+ ALL.49–51 A primary concern with the tyrosine kinase inhibitors is the emergence of resistance, specifically T315I mutations. Ponatinib is the only BCR-ABL tyrosine kinase inhibitor available in the United States with known activity against T315I mutations.52 The use of the BCR-ABL tyrosine kinase inhibitors other than imatinib for first-line therapy is not well established.
In adults with B-cell ALL, about 50% have leukemic cells that express CD20. CD20 expression has been associated with decreased CR rates and OS.53 A phase II study has evaluated hyperCVAD and rituximab versus hyperCVAD alone and reported a higher CR rate (70% vs. 38%) and longer OS (75% vs. 47%) in patients treated with hyperCVAD and rituximab.54 These results are encouraging and support the use of rituximab in patients who have cells that express CD20.
HSCT plays an important role in the treatment of adult patients with ALL. For patients with Ph+ ALL who have a CR after induction therapy, consolidation with allogeneic HSCT should be considered if a human leukocyte antigen (HLA)-matched sibling or matched unrelated donor is available. After HSCT, patients should continue with standard maintenance therapy that includes a tyrosine kinase inhibitor. For patients with Philadelphia chromosome negative (Ph–) disease who have MRD after induction therapy or have high-risk features for relapse, allogeneic HSCT should be considered if a matched donor is available.55
Relapsed Acute Lymphoblastic Leukemia
About 20% of children with ALL will relapse.56 The most common site for relapse is the bone marrow (53%), although isolated relapses can occur in the CNS (19%) or testicles (5%), in addition to multiple sites of disease.57 Because marrow relapse usually follows isolated CNS or testicular relapses, patients with isolated extramedullary relapses are treated with localized radiation (cranial or testicular) and aggressive systemic chemotherapy similar to that given to patients with a marrow relapse.58
Children who fail to achieve a CR by day 29 of initial remission induction therapy usually receive an additional 2 weeks of four-drug induction therapy. If the children do not achieve a CR with the additional therapy, they are usually treated for bone marrow relapse consisting of intensive induction therapy consisting of at least three cycles of chemotherapy. Examples of regimens include vincristine, pegaspargase, corticosteroid, and doxorubicin; etoposide, cyclophosphamide, and high-dose methotrexate; and high-dose cytarabine and asparaginase.
Patients who have completed treatment and have stayed in remission for longer periods are more likely to be reinduced into remission again. Patients with more favorable risk factors initially, and those who received less intensive initial treatments, are more likely to respond well to reinduction/salvage regimens. The second CR rate is 78% in children who were in continuous complete remission for less than 18 months, 78% if the duration of remission was 18 to 36 months, and 93% if the duration of remission was more than 36 months.56 Three-year OS following bone marrow (28%), CNS (60%), and testicular (60%) relapse is not optimal.58 Overall, 5-year disease-free survival rates are 27% for second complete remission (CR2) and 15% for third complete remission (CR3).56 Clofarabine, a purine antimetabolite, is an option for patients on second or later relapses, but the duration of response is less than 6 months. Current trials are evaluating the place for clofarabine in combination with other agents.
Allogeneic HSCT has traditionally been the treatment of choice for early bone marrow relapse (continuous CR less than 36 months) while children who relapse more than 36 months after completion of initial therapy have traditionally received chemotherapy alone.22 Some more recent analyses have shown HSCT to be an advantage to all relapsed children, while some have not shown a benefit.56 Therefore, the question of who would benefit from HSCT continues to be investigated.
Most patients with relapsed or refractory disease are considered for an allogeneic HSCT with a matched sibling or unrelated donor if they achieve a CR2 following salvage chemotherapy. Most elderly patients are not candidates for standard allogeneic HSCT but are candidates for nonmyeloablative transplant (NMT). Patients who undergo a NMT receive a reduced intensity conditioning regimen. A NMT may produce similar outcomes with less treatment-related morbidity and mortality. The National Marrow Donor Program and the American Society for Blood and Marrow Transplantation have developed guidelines for transplant consultation based on current clinical practice and evidence-based medicine.55,59
For relapsed or refractory disease in Ph+ patients, leukemic cells should be tested for mutations to guide selection of the tyrosine kinase inhibitor. In addition, nelarabine may be an option for adults with relapsed or refractory T-cell disease.
Late Effects of Treatment
Certain late effects associated with cranial or craniospinal irradiation and corticosteroids were discussed earlier. The Childhood Cancer Survivor Study tracks the health status of adults treated for childhood cancer between 1970 and 1986 and has yielded invaluable information on how to monitor adult survivors.60 Leukemia survivors are 3.7 times more likely to develop a severe or life-threatening chronic health condition as compared with healthy siblings, and 2.8 times more likely to report multiple chronic conditions.60
Older ALL regimens that incorporated intensive use of topoisomerase II inhibitors (etoposide and teniposide) are associated with unacceptably high risks of development of secondary leukemia.22 High cumulative doses of anthracyclines used in high-risk or relapsed patients can cause cardiomyopathy. Cranial irradiation is also associated with learning deficits, especially in patients younger than 5 years of age at the time of treatment. Patients who received cranial radiation as children also have higher unemployment rates and lower marital rates among females two decades after diagnosis.60 The Children’s Oncology Group has developed long-term follow-up guidelines for survivors of childhood, adolescent, and young adult cancers (www.survivorshipguidelines.org).
ACUTE MYELOID LEUKEMIA
Risk Classification
Many clinical and laboratory features at diagnosis are associated with response to treatment, as measured by the CR rate, duration of remission, and long-term survival. Identification of these risk factors may allow the clinician to better understand the disease and to tailor treatment according to risk of disease recurrence. For example, if a patient has many clinical and laboratory features that are associated with a good response to chemotherapy (“good risk”), then the clinician may choose to give less intensive therapy to reduce the risk of long-term toxic effects. Conversely, if a patient is unlikely to respond well to therapy (“high risk”), then the clinician may choose to give more intensive chemotherapy that may include HSCT.
Several prognostic factors have been identified for adults with AML. ![]() The most important patient factor is age, with younger patients more likely to achieve a CR than patients older than age 60 years.3,61 The lower CR rate in older patients results from an increased frequency of fatal infection and bleeding complications and resistance to conventional chemotherapy. The duration of remission is also shorter in older patients as compared to younger patients. Other patient-specific prognostic factors include concurrent infection and any major organ impairment.3 Patients with extramedullary disease, CNS involvement, or underlying MDS have a worse prognosis. Other unfavorable prognostic factors in adult AML include: age older than 60 years, multidrug-resistance gene expression, WBC >100,000 cells/mm3(100 × 109/L), and therapy-related AML.62 Age must be evaluated as a continuous variable when looking at prognostic factors. The clinical difference between a patient 61 years old and one 71 years old, is much greater than a 59-year-old and a 61-year-old. Certain cytogenetic abnormalities are also known to worsen the response rate and survival of patients with AML (Table 111-4).3,63 Chromosome 16 or translocations between chromosome 8 and 21 alter core-binding factor. Core-binding factor is associated with sensitivity to cytarabine.64 In addition, patients who develop a “secondary” leukemia after treatment of another malignancy usually have a very poor response to antileukemic chemotherapy (i.e., therapy-related AML). Another factor that needs consideration for any cancer treatment is performance status. A bedridden patient with a new diagnosis of AML would not be a good candidate for treatment because of high treatment-related morbidity and mortality. Patients with poor performance status may be offered supportive care.
The most important patient factor is age, with younger patients more likely to achieve a CR than patients older than age 60 years.3,61 The lower CR rate in older patients results from an increased frequency of fatal infection and bleeding complications and resistance to conventional chemotherapy. The duration of remission is also shorter in older patients as compared to younger patients. Other patient-specific prognostic factors include concurrent infection and any major organ impairment.3 Patients with extramedullary disease, CNS involvement, or underlying MDS have a worse prognosis. Other unfavorable prognostic factors in adult AML include: age older than 60 years, multidrug-resistance gene expression, WBC >100,000 cells/mm3(100 × 109/L), and therapy-related AML.62 Age must be evaluated as a continuous variable when looking at prognostic factors. The clinical difference between a patient 61 years old and one 71 years old, is much greater than a 59-year-old and a 61-year-old. Certain cytogenetic abnormalities are also known to worsen the response rate and survival of patients with AML (Table 111-4).3,63 Chromosome 16 or translocations between chromosome 8 and 21 alter core-binding factor. Core-binding factor is associated with sensitivity to cytarabine.64 In addition, patients who develop a “secondary” leukemia after treatment of another malignancy usually have a very poor response to antileukemic chemotherapy (i.e., therapy-related AML). Another factor that needs consideration for any cancer treatment is performance status. A bedridden patient with a new diagnosis of AML would not be a good candidate for treatment because of high treatment-related morbidity and mortality. Patients with poor performance status may be offered supportive care.
TABLE 111-4 Risk Category According to Cytogenetic and Molecular Abnormalities Present




