Acute Kidney Injury
KEY CONCEPTS
![]() Three classification systems exist for staging severity of acute kidney injury (AKI): (a) Risk, Injury, Failure, Loss of Kidney Function, and End-Stage Kidney Disease (RIFLE), (b) Acute Kidney Injury Network (AKIN), and (c) Kidney Disease: Improving Global Outcomes (KDIGO) clinical practice guidelines. All three classification systems are based on separate criteria for serum creatinine (Scr) and urine output.
Three classification systems exist for staging severity of acute kidney injury (AKI): (a) Risk, Injury, Failure, Loss of Kidney Function, and End-Stage Kidney Disease (RIFLE), (b) Acute Kidney Injury Network (AKIN), and (c) Kidney Disease: Improving Global Outcomes (KDIGO) clinical practice guidelines. All three classification systems are based on separate criteria for serum creatinine (Scr) and urine output.
![]() AKI is a common complication in hospitalized patients and is associated with high morbidity and mortality, especially in critically ill.
AKI is a common complication in hospitalized patients and is associated with high morbidity and mortality, especially in critically ill.
![]() AKI is categorized based on three distinct types of injury: (a) prerenal—decreased renal blood flow, (b) intrinsic—structural damage within the kidney, and (c) postrenal—an obstruction is present within the urine collection system.
AKI is categorized based on three distinct types of injury: (a) prerenal—decreased renal blood flow, (b) intrinsic—structural damage within the kidney, and (c) postrenal—an obstruction is present within the urine collection system.
![]() Conventional formulas used to determine estimated glomerular filtration rate (eGFR) and creatinine clearance should not be used to estimate renal function in patients with AKI. This may be especially true for medication dosing adjustments.
Conventional formulas used to determine estimated glomerular filtration rate (eGFR) and creatinine clearance should not be used to estimate renal function in patients with AKI. This may be especially true for medication dosing adjustments.
![]() Prevention is of utmost importance since there are very few therapeutic options available for the treatment of established AKI.
Prevention is of utmost importance since there are very few therapeutic options available for the treatment of established AKI.
![]() Supportive management remains the primary approach to prevent or reduce the complications associated with AKI. Supportive therapies include renal replacement therapy (RRT), nutritional support, avoidance of nephrotoxins, and blood pressure and fluid management.
Supportive management remains the primary approach to prevent or reduce the complications associated with AKI. Supportive therapies include renal replacement therapy (RRT), nutritional support, avoidance of nephrotoxins, and blood pressure and fluid management.
![]() For those patients with prolonged or severe AKI, RRT is the cornerstone of support along with an aggressive approach to fluid, electrolyte, and waste management.
For those patients with prolonged or severe AKI, RRT is the cornerstone of support along with an aggressive approach to fluid, electrolyte, and waste management.
![]() Drug dosing for AKI patients receiving continuous renal replacement therapy (CRRT) or sustained low-efficiency dialysis (SLED) is poorly characterized. Dosing regimens should be individualized and therapeutic drug monitoring utilized whenever possible.
Drug dosing for AKI patients receiving continuous renal replacement therapy (CRRT) or sustained low-efficiency dialysis (SLED) is poorly characterized. Dosing regimens should be individualized and therapeutic drug monitoring utilized whenever possible.
![]() Diuretic resistance is a common phenomenon in the patient with AKI and can be addressed with sodium restriction, combination diuretic therapy, or a continuous infusion of a loop diuretic.
Diuretic resistance is a common phenomenon in the patient with AKI and can be addressed with sodium restriction, combination diuretic therapy, or a continuous infusion of a loop diuretic.
INTRODUCTION
Acute kidney injury (AKI) is a clinical syndrome generally defined by an abrupt reduction in kidney function as evidenced by changes in laboratory values, serum creatinine (Scr), blood urea nitrogen (BUN), and urine output. The consequences of AKI can be serious, especially in hospitalized patients, among whom complications and mortality are particularly high. Early recognition along with supportive therapy is the focus of management for those with established AKI, as there is no therapy that directly reverses the injury. Individuals at risk, such as those with history of chronic kidney disease (CKD), need to have their hemodynamic status carefully monitored and their exposure to nephrotoxins minimized. A thorough patient workup is often necessary and includes past medical and surgical history, medication use, physical examination, and multiple laboratory tests. Management goals include maintenance of blood pressure, fluid, and electrolyte homeostasis, all of which may be dramatically altered. Additional therapies designed to eliminate or minimize the insult that precipitated AKI include discontinuation of the offending drug (i.e., the nephrotoxin), aggressive hydration, maintenance of renal perfusion, and renal replacement therapy (RRT).
In this chapter, the definition, classification, epidemiology, and common etiologies of AKI are presented. Methods to recognize and assess the extent of kidney function loss are also discussed. Finally, preventive strategies for patients at risk and management approaches for those with established AKI are reviewed.
DEFINITION AND CLASSIFICATION OF ACUTE KIDNEY INJURY
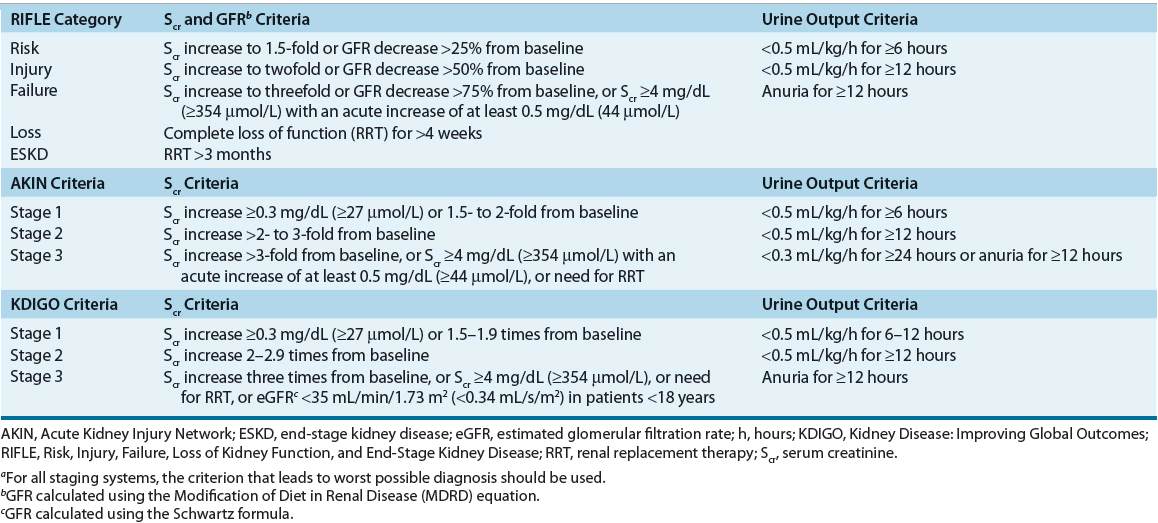
![]() Over the past 10 years, several efforts by a broad consensus of experts have been made to standardize the definition and classification of AKI. In 2004, the Acute Dialysis Quality Initiative (ADQI) group published a consensus-derived definition and classification system called the Risk, Injury, Failure, Loss of Kidney Function, and End-Stage Kidney Disease (RIFLE) classification.1 In 2007, a modified version of RIFLE was developed by the Acute Kidney Injury Network (AKIN) and these criteria are presented in Table 28-12 (see Table 28-1 for an overview of all classification systems). Both classification systems are now widely accepted and have been validated to predict outcomes in thousands of patients worldwide.3,4 While generally similar, there are a few noteworthy differences: RIFLE defines AKI as an abrupt (1 to 7 days) but sustained (>24 hours) decrease in renal function from baseline while AKIN designates a 48-hour period for the decrease to occur. Also, AKIN removed RIFLE’s last two classification components (Loss of Kidney Function and End-Stage Kidney Disease [ESKD]) from the staging system and instead places all patients receiving RRT automatically into AKIN stage 3. Finally, AKIN removed all estimated glomerular filtration rate (eGFR) criteria from its staging system and lowered the absolute increase in Scr from 0.5 mg/dL (44 μmol/L) designated for RIFLE-Risk class to 0.3 mg/dL (27 μmol/L) for AKIN stage 1.1,2
Over the past 10 years, several efforts by a broad consensus of experts have been made to standardize the definition and classification of AKI. In 2004, the Acute Dialysis Quality Initiative (ADQI) group published a consensus-derived definition and classification system called the Risk, Injury, Failure, Loss of Kidney Function, and End-Stage Kidney Disease (RIFLE) classification.1 In 2007, a modified version of RIFLE was developed by the Acute Kidney Injury Network (AKIN) and these criteria are presented in Table 28-12 (see Table 28-1 for an overview of all classification systems). Both classification systems are now widely accepted and have been validated to predict outcomes in thousands of patients worldwide.3,4 While generally similar, there are a few noteworthy differences: RIFLE defines AKI as an abrupt (1 to 7 days) but sustained (>24 hours) decrease in renal function from baseline while AKIN designates a 48-hour period for the decrease to occur. Also, AKIN removed RIFLE’s last two classification components (Loss of Kidney Function and End-Stage Kidney Disease [ESKD]) from the staging system and instead places all patients receiving RRT automatically into AKIN stage 3. Finally, AKIN removed all estimated glomerular filtration rate (eGFR) criteria from its staging system and lowered the absolute increase in Scr from 0.5 mg/dL (44 μmol/L) designated for RIFLE-Risk class to 0.3 mg/dL (27 μmol/L) for AKIN stage 1.1,2
TABLE 28-1 RIFLE, AKIN, and KDIGO Classification Schemes for Acute Kidney Injurya
Even though the initial aim of RIFLE and the AKIN modification was to provide a standardized definition of AKI, they resulted in two distinct definitions that were not consistently applied across studies and thus have provided somewhat different epidemiologic findings. In order to provide a single definition of AKI for practice, research, and public health, a second modification of RIFLE and AKIN criteria was recently published by the Kidney Disease: Improving Global Outcomes (KDIGO) Clinical Practice Guidelines working group in 2012.5
KDIGO defines AKI as being present if any of the following three criteria are met: 1. Increase in Scr by at least 0.3 mg/dL (27 μmol/L) within 48 hours, 2. Increase in Scr by at least 1.5 times baseline within the prior 7 days, or 3. Decrease in urine volume to less than 0.5 mL/kg/h for 6 hours.
KDIGO staging of AKI is similar to the RIFLE and AKIN criteria with the notable addition of inclusion of pediatric patients (<18 years) to KDIGO Stage 3 for those with an estimated GFR of less than 35 mL/min/1.73 m2 (0.34 mL/s/m2) as determined by the Schwartz formula.5 Due to the very recent publication of KDIGO guidelines, it still remains to be seen if it will supersede RIFLE and AKIN criteria for the diagnosis and classification of AKI in the future.
Since all three staging systems depend on Scr and urine output as the main diagnostic criteria, they are associated with the same inherent weaknesses. An increase in Scr is usually evident about 1 or 2 days after development of AKI. This lag time in Scr rise may significantly delay diagnosis of AKI and adversely affect patient outcomes. Urine output reduction emerges earlier in AKI but is a very nonspecific marker because it may not always be present. In fact, patients with AKI can be anuric (urine output <50 mL/day), oliguric (urine output <500 mL/day), or nonoliguric (urine output >500 mL/day). Urine output will also vary with volume status, diuretic administration, and presence of obstruction.6 Further, since all criteria are based on detecting a decrease in Scr from its baseline, a patient’s renal function prior to the development of AKI needs to be known. If the baseline measure of Scr is not available and the patient has no history of renal dysfunction, the ADQI, a workgroup composed of experts in nephrology and critical care, has suggested estimating the baseline Scr value by using the four variable Modification of Diet in Renal Disease (MDRD) equation with an assumed normal GFR of 75 mL/min/1.73 m2 (0.72 mL/s/m2).1 However, this method needs to be interpreted with caution as it has been found to overestimate the incidence of AKI by as much as 40%.7,8
EPIDEMIOLOGY
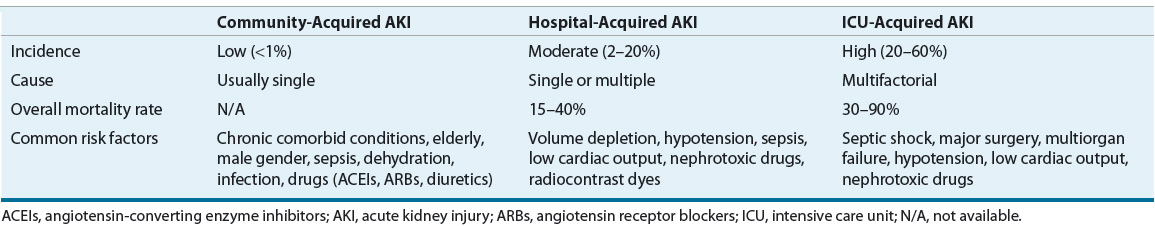
The epidemiology of AKI varies widely depending on the patient population, geographical location, and the criteria used to evaluate the patient. AKI is generally considered to be an uncommon condition in the community-dwelling population, with an annual incidence of 520 per 100,000 person-years for nondialysis requiring AKI and 30 per 100,000 person-years for dialysis-requiring injury9 (Table 28-2). AKI is more common in hospitalized individuals, with a reported incidence ranging from 2% to 20%.10,11 Intensive care unit (ICU) patients have the highest risk of developing AKI, with 20% to 60% of critically ill patients being affected.3,4
TABLE 28-2 Incidence and Outcomes of AKI
![]() Increased mortality and morbidity are two well-recognized complications of AKI. In particular, severity, duration, and frequency of AKI appear to be important predictors of poor patient outcomes. Any degree of AKI is associated with an increased risk of death, and the odds increase with the severity of the insult.11,12 For survivors of AKI, the development of some degree of CKD and need for RRT are other important considerations.13 In addition, AKI is associated with increased length of hospital stay, ventilator days, and need for posthospitalization care.10,13
Increased mortality and morbidity are two well-recognized complications of AKI. In particular, severity, duration, and frequency of AKI appear to be important predictors of poor patient outcomes. Any degree of AKI is associated with an increased risk of death, and the odds increase with the severity of the insult.11,12 For survivors of AKI, the development of some degree of CKD and need for RRT are other important considerations.13 In addition, AKI is associated with increased length of hospital stay, ventilator days, and need for posthospitalization care.10,13
ETIOLOGY
![]() The etiology of AKI can be divided into broad categories based on the anatomic location of the injury associated with the precipitating factor(s). The management of patients presenting with this disorder is largely predicated on identification of the specific etiology responsible for the patient’s AKI (Fig. 28-1). Traditionally, the causes of AKI have been categorized as (a) prerenal, which results from decreased renal perfusion in the setting of undamaged parenchymal tissue, (b) intrinsic, the result of structural damage to the kidney, most commonly the tubule from an ischemic or toxic insult, and (c) postrenal, caused by obstruction of urine flow downstream from the kidney (Fig. 28-2).
The etiology of AKI can be divided into broad categories based on the anatomic location of the injury associated with the precipitating factor(s). The management of patients presenting with this disorder is largely predicated on identification of the specific etiology responsible for the patient’s AKI (Fig. 28-1). Traditionally, the causes of AKI have been categorized as (a) prerenal, which results from decreased renal perfusion in the setting of undamaged parenchymal tissue, (b) intrinsic, the result of structural damage to the kidney, most commonly the tubule from an ischemic or toxic insult, and (c) postrenal, caused by obstruction of urine flow downstream from the kidney (Fig. 28-2).
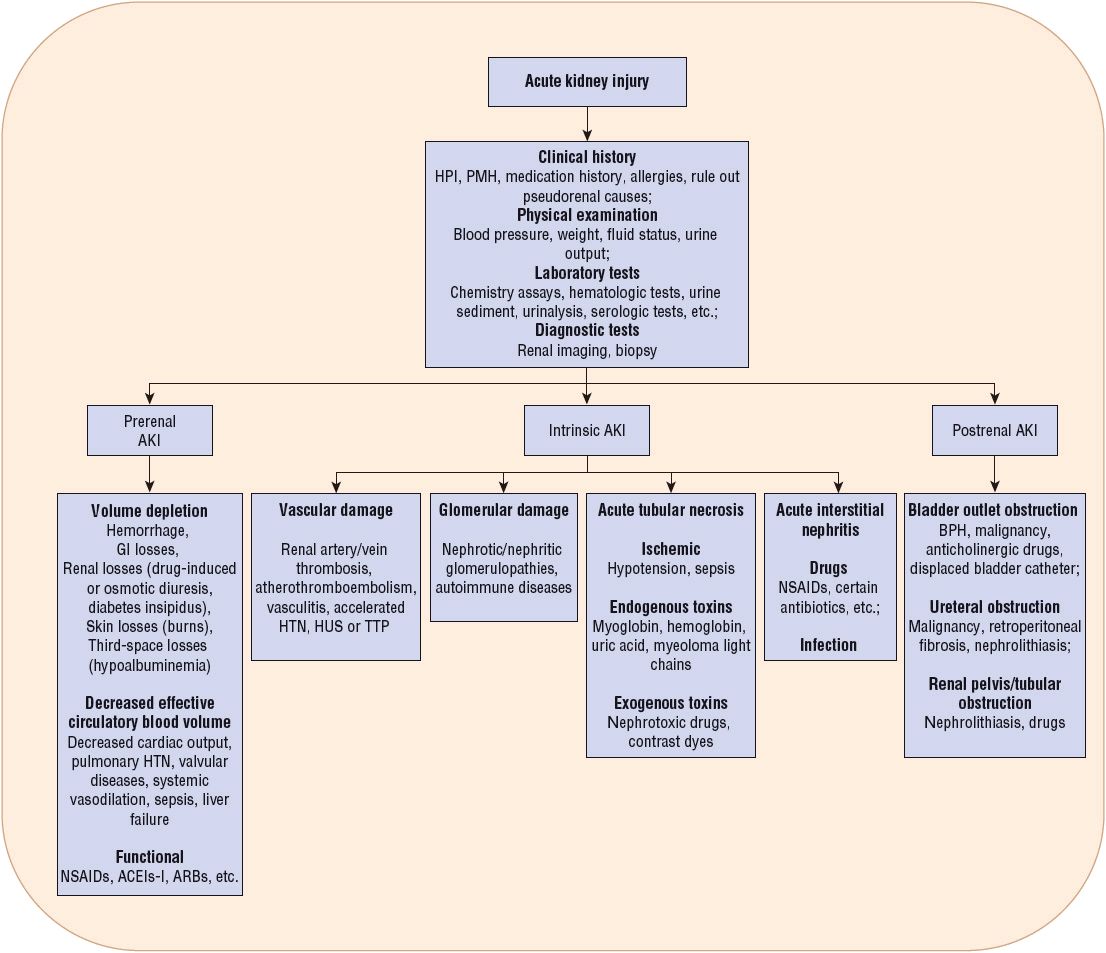
FIGURE 28-1 Classification of acute kidney injury (AKI) based on etiology. (ACEIs, angiotensin-converting enzyme inhibitors; ARBs, angiotensin receptor blockers; BPH, benign prostatic hyperplasia; HPI, history of present illness; HTN, hypertension; HUS, hemolytic uremic syndrome; NSAIDs, nonsteroidal antiinflammatory drugs; PMH, past medical history; TTP, thrombotic thrombocytopenic purpura.)
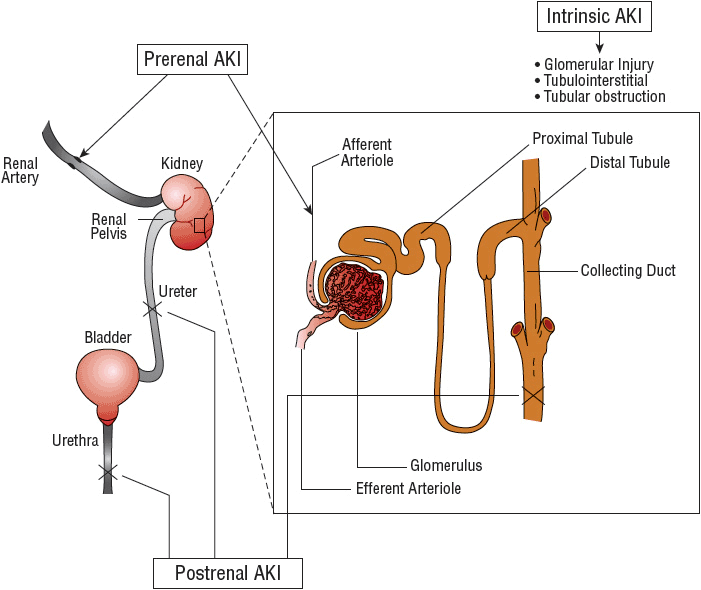
FIGURE 28-2 Physiologic classification of AKI. Blood flows through the afferent arteriole, to the glomerulus, and exits through the efferent arteriole. A decrease in blood flow and renal perfusion can lead to a prerenal reduction in renal function. Under conditions in which renal blood flow is diminished, the kidney maintains glomerular ultrafiltration by vasodilating the afferent arterioles and vasoconstricting the efferent arterioles. Medications that may interfere with these processes may result in an abrupt decline in glomerular filtration. Damage to the glomerular or tubular regions leads to intrinsic AKI. Obstruction of urine flow in the collecting tubule, ureter, bladder, or urethra is termed postrenal impairment.
Community-acquired AKI most commonly occurs secondary to renal hypoperfusion from volume depletion (dehydration, vomiting, and diarrhea), sepsis, or medications (angiotensin-converting enzyme inhibitors [ACEIs], angiotensin receptor blockers [ARBs], and diuretics).9,14,15 The most common cause of hospital-and ICU-acquired AKI is intrinsic, occurring as the result of acute tubular necrosis (ATN).
The risk of AKI increases substantially with decreasing GFR and presence of underlying CKD.16 Other risk factors for developing AKI are age >65 years, septic shock, critical illness, chronic diseases (heart, lung, liver), recent exposure to nephrotoxic drugs, cardiac surgery, cancer, trauma, and African American race.5,15,17 History of AKI has also been associated with high risk for developing additional episodes of AKI and subsequent complications such as advanced CKD.18
PATHOPHYSIOLOGY
The three main pathophysiologic processes involved in the development of AKI include prerenal AKI, intrinsic AKI, and postrenal AKI. As described below, pseudorenal kidney injury does not represent a true pathophysiologic process.
Pseudorenal Acute Kidney Injury
Pseudorenal AKI is characterized by a rise in either the BUN or the Scr, which misleadingly may suggest the presence of renal dysfunction, when in fact GFR is not diminished. This could be the result of cross-reactivity with the assay used to measure the BUN or Scr or selective inhibition of the secretion of creatinine into the proximal tubular lumen by certain medications (see eChap. 17). A similar problem exists when urine output data are unreliable. Urine output may be either inaccurate (particularly in noncatheterized patients) or not reported at all. Since the urine output criteria for AKI staging are weight-based, some obese individuals may meet the definition of AKI without truly having any kidney impairment. Thus, clinical judgment should always be applied when interpreting laboratory results.
Prerenal Acute Kidney Injury
Prerenal AKI or prerenal azotemia results from hypoperfusion of the renal parenchyma, with or without systemic arterial hypotension. Renal hypoperfusion with systemic arterial hypotension may be caused by a decline in either the intravascular volume or the effective circulating blood volume. Intravascular volume depletion may result from several conditions, including hemorrhage, excessive GI losses (severe vomiting or diarrhea), dehydration, extensive burns, and diuretic therapy. Effective circulating blood volume may be reduced in conditions associated with a decreased cardiac output and systemic vasodilation (e.g., sepsis). Renal hypoperfusion without systemic hypotension is most commonly associated with bilateral renal artery occlusion or unilateral occlusion in a patient with a single functioning kidney.
Patients with a mild reduction in effective circulating blood volume or volume depletion are generally able to maintain a normal GFR by activating several compensatory mechanisms. Those initial physiologic responses by the body stimulate the sympathetic nervous and the renin–angiotensin–aldosterone system and release antidiuretic hormone if hypotension is present. These responses work together to directly maintain blood pressure via vasoconstriction and stimulation of thirst, which in conscious patients results in increased fluid intake, as well as sodium and water retention. Additionally, GFR may be maintained by afferent arteriole dilation (mediated by intrarenal production of vasodilatory prostaglandins, kallikrein, kinins, and nitric oxide) and efferent arteriole constriction (mainly mediated by angiotensin II). In concert, these homeostatic mechanisms are often able to maintain arterial pressure and renal perfusion, potentially averting the progression to AKI.19 If, however, the decreased renal perfusion is severe or prolonged, these compensatory mechanisms may be overwhelmed, and prerenal AKI will be clinically evident.
Patients at risk for prerenal AKI are particularly susceptible to changes in the afferent and efferent arteriolar tone, as they may not be able to compensate as readily. Certain drug classes can interfere with these renal adaptive responses that are normally responsible for maintaining adequate renal perfusion. The resulting reduction in the glomerular hydrostatic pressure precipitates an abrupt decline in GFR and is sometimes referred to as functional AKI. A common cause of this syndrome is a decrease in efferent arteriolar resistance as the result of initiation of an ACE inhibitor or ARB (see Chap. 31). For example, individuals with heart failure are often given an ACE inhibitor or ARB to help improve left ventricular function, but if the dose is titrated too rapidly, they may experience a decline in GFR. If the increase in the Scr is less than 30% from baseline, the medication can generally be continued. Another classic example is initiation of ACE inhibitors or ARBs in patients with renovascular disease. It is estimated that ACE inhibitor–induced renal failure occurs in 6% to 23% of patients with bilateral renal artery stenosis and in 38% of patients with unilateral stenosis who have a single kidney.20 As a result, administration of ACE inhibitor or ARB therapy in the presence of those conditions is contraindicated. Nonsteroidal antiinflammatory drugs (NSAIDs) may also initiate AKI in susceptible individuals. NSAIDs inhibit renal prostaglandin production and afferent arteriolar vasodilation, which some patients rely on to maintain renal perfusion and GFR. Patients at risk for NSAID-induced AKI include those with CKD, volume depletion, and decreased effective circulating blood volume.21
If the causes of renal hypoperfusion are promptly corrected, prerenal AKI can be reversed and renal function returned to baseline in a matter of days. Prolonged prerenal azotemia, in contrast, can cause direct (and potentially irreversible) injury to the renal parenchyma and lead to development of ischemic ATN.22
Intrinsic Acute Kidney Injury
Intrinsic AKI results from direct damage to the kidney and is categorized on the basis of the injured structures within the kidney: the renal vasculature, glomeruli, tubules, and interstitium.
Renal Vasculature Damage
Occlusion of the larger renal vessels resulting in AKI is not common but can occur if large atheroemboli or thromboemboli occlude the bilateral renal arteries or one vessel of the patient with a single kidney. Atheroemboli most commonly develop during vascular procedures that cause atheroma dislodgement, such as angioplasty and aortic manipulations. Thromboemboli may arise from dislodgement of a mural thrombus in the left ventricle of a patient with severe heart failure or from the atria of a patient with atrial fibrillation. Renal artery thrombosis may occur in a similar fashion to coronary thrombosis, in which a thrombus forms in conjunction with an atherosclerotic plaque.
Although smaller vessels can also be obstructed by atheroemboli or thromboemboli, the damage is limited to the vessels involved, and the development of significant AKI is unlikely. However, these small vessels are susceptible to inflammatory processes that lead to microvascular damage and vessel dysfunction when the renal capillaries are affected. Neutrophils invade the vessel wall, causing damage that can include thrombus formation, tissue infarction, and collagen deposition within the vessel structure. Diffuse renal vasculitis can be mild or severe, with severe forms promoting concomitant ischemic ATN. The Scr is usually elevated when the lesions are diffuse. Accelerated hypertension that is not treated may also compromise renal microvascular blood flow, causing diffuse renal capillary damage.
Glomerular Damage
Only 5% of the cases of intrinsic AKI are of glomerular origin. The glomerulus is one of two capillary beds in the kidney. It serves to filter fluid and solute into the tubules while retaining proteins and other large blood components in the intravascular space. Because the glomerulus is a capillary system, similar damage observed in the renal vasculature can additionally occur by the same mechanisms. The pathophysiology and specific therapeutic approaches to glomerulonephritis are described in detail in Chapter 32.
Tubular Damage
Approximately 85% of all cases of intrinsic AKI are caused by ATN, of which 50% are a result of renal ischemia, often arising from an extended prerenal state. The remaining 35% are the result of exposure to direct tubule toxins, which can be endogenous (myoglobin, hemoglobin, or uric acid) or exogenous (contrast agents, aminoglycosides, etc.). The tubules located within the medulla of the kidney are particularly at risk for ischemic injury, as this portion of the kidney is metabolically active and thus has high oxygen requirements, yet, as compared with the cortex, receives relatively low oxygen delivery. Thus, ischemic conditions caused by severe hypotension or exposure to vasoconstrictive drugs preferentially affect the tubules more than any other portion of the kidney.
The clinical evolution of ATN is characterized by three distinct phases: initiation, maintenance, and recovery. The hallmarks of the initiation phase are ischemic injury and GFR reduction, both of which occur as a result of the interplay between several different pathophysiologic processes. Ischemic injury causes tubular epithelial cell necrosis or apoptosis and is followed by an extension phase with continued hypoxia and an inflammatory response involving the nearby interstitium. The loss of epithelial cells between the filtrate and the interstitium leaves the basement membrane denuded and unable to appropriately regulate fluid and electrolyte transfer across the tubular lumen. As a result, the glomerular filtrate starts leaking back into the interstitium and is reabsorbed into the systemic circulation. Additionally, urine flow is obstructed by accumulation of sloughed epithelial cells, cellular debris, and formation of casts. The onset of ATN can occur over hours to days, depending on the factors responsible for the damage. Regardless of the etiology, tubular injury, back leakage, and obstruction lead to decreased urine-concentrating ability, decreased urine output, and, ultimately, reduced GFR. Continued kidney hypoxia or toxin exposure after the original insult kills more cells and propagates the inflammatory response. It also can extend the injury and delay the recovery process. With prolonged ischemia, the tubular epithelial cells in the corticomedullary junction are damaged and die.22 When the toxin or ischemia is removed, a maintenance phase ensues and may last anywhere from a few weeks to several months. The maintenance phase is eventually followed by a recovery phase, during which new tubule cells are regenerated. The recovery phase is associated with a notable diuresis, which requires prompt attention to maintain fluid balance, or a secondary prerenal injury may occur. However, if the ischemia or injury is extremely severe or prolonged, cortical necrosis may occur, limiting tubule cell regrowth in the affected areas.20
Interstitial Damage
If the renal interstitium becomes severely inflamed and edematous, it can lead to development of acute interstitial nephritis (AIN). AIN may be caused by drugs (see Chap. 31), infections, and, rarely, autoimmune idiopathic diseases. Whatever the inciting event, acute interstitial injury is characterized by lesions composed of monocytes, eosinophils, macrophages, B cells, or T cells, clearly identifying an immunologic response as the injurious process affecting the interstitium. If AIN is caused by a drug hypersensitivity reaction, most patients will regain normal renal function within several weeks if the offending drug is promptly discontinued. If symptoms of AIN remain unrecognized, and the exposure to the causative agent continues, persistent renal dysfunction associated with interstitial fibrosis and tubular atrophy may develop.23
Postrenal Acute Kidney Injury
Postrenal AKI accounts for less than 5% of all cases of AKI and may develop as the result of obstruction at any level within the urinary collection system22 (see Fig. 28-1). However, if the obstructing process is above the bladder, it must involve both kidneys (one kidney in a patient with a single functioning kidney) to cause clinically significant AKI, as one functioning kidney can generally maintain a near-normal GFR. Bladder outlet obstruction, the most common cause of obstructive uropathy, is often the result of a prostatic process (hypertrophy, cancer, or infection), producing a physical impingement on the urethra and thereby preventing the passage of urine. It may also be the result of an improperly placed urinary catheter. Blockage may also occur at the ureter level secondary to nephrolithiasis, blood clots, sloughed renal papillae, or physical compression by an abdominal process. Crystal deposition within the tubules from oxalate and some medications severe enough to cause AKI is uncommon, but it is possible in patients with severe volume contraction and in those receiving large doses of a drug with relatively low urine solubility (see Chap. 31). In these cases, patients have insufficient urine volume to prevent crystal precipitation in the urine. Extremely elevated uric acid concentrations from chemotherapy-induced tumor lysis syndrome can cause obstruction and direct tubular injury as well.24 Wherever the location of the obstruction, urine will accumulate in the renal structures above the obstruction and cause increased pressure upstream. The ureters, renal pelvis, and calyces all expand, and the net result is a decline in GFR. If renal vasoconstriction ensues, a further decrement in GFR will be observed.
CLINICAL PRESENTATION
The initiating signs or symptoms prompting the clinical suspicion of AKI is highly variable and largely dependent on the underlying etiology. It may be a change in urinary habits (e.g., decreased urine output or urine discoloration), sudden weight gain, or severe abdominal or flank pain. Early recognition and cause identification are critical, as they directly affect the outcome of AKI. One of the first steps in the diagnostic process is to determine if the renal complication is acute, chronic, or the result of an acute change in a patient with known CKD (also called acute-on-chronic renal failure). Patients should also be promptly evaluated for any changes in their fluid and electrolyte status. Patients presenting with AKI in the outpatient environment may have very nonspecific or seemingly unrelated symptoms so that the time of onset of the injury can be difficult to determine. On the other hand, AKI in hospitalized patients is often detected much earlier in its course due to frequent laboratory studies and daily patient assessment.
Patient Assessment
The assessment of a patient with AKI starts with a thorough review of his or her medical records, with a particular focus on chronic conditions, medication history, laboratory studies, procedures, and surgeries. An exhaustive review of prescription and nonprescription medicines, herbal products, and recreational drugs may help determine if AKI was potentially precipitated by drug ingestion.
During the initial patient evaluation, presumptive signs and symptoms of AKI need to be differentiated from a potential new diagnosis of CKD. A past medical history for renal disease–related chronic conditions (e.g., poorly controlled hypertension and diabetes mellitus), previous laboratory data documenting the presence of proteinuria or an elevated Scr, and the finding of bilateral small kidneys on renal ultrasonography suggest the presence of CKD rather than AKI. However, it is important to note that patients with CKD may develop episodes of AKI as well. In that case, an abrupt rise in the patient’s baseline Scr is one of the most useful indicators of the presence of an acute insult to the kidneys.
An acute change in urinary habits is another common and noticeable symptom associated with AKI. The presence of cola-colored urine is indicative of blood in the urine, a finding commonly associated with acute glomerulonephritis. In hospitalized patients, changes in urine output may be helpful in characterizing the cause of the patient’s AKI. Acute anuria is typically caused by either complete urinary obstruction or a catastrophic event (e.g., shock or acute cortical necrosis). Oliguria, which often develops over several days, suggests prerenal azotemia, whereas nonoliguric renal failure usually results from acute intrinsic renal failure or incomplete urinary obstruction.
Depending on the underlying cause of AKI, patients may present with a variety of symptoms affecting virtually any organ system of the body. Constitutional symptoms such as nausea, vomiting, fatigue, malaise, and weight gain are common but nonspecific. The onset of flank pain is suggestive of a urinary stone; however, if bilateral, it may suggest swelling of the kidneys secondary to acute glomerulonephritis or AIN. Complaints of severe headaches may suggest the presence of severe hypertension and vascular damage. The presence of fever, rash, and arthralgias may be indicative of drug-induced AIN or lupus nephritis.
A thorough physical examination is an important step in evaluating individuals with AKI, as clues regarding the etiology can be evident from the patient’s head (eye examination) to toe (evidence of dependent edema) assessment. Observations will either support or refute the cause as prerenal, intrinsic, or postrenal. Evaluation of the patient’s volume and hemodynamic status is critical as well, as it will guide management. For example, patients with prerenal AKI can present with either volume depletion or fluid overload. Volume depletion may be evidenced by the presence of postural hypotension, decreased jugular venous pressure (JVP), and dry mucous membranes. Fluid overload, on the other hand, is often reflected by elevated JVP, pitting edema, ascites, and pulmonary crackles.
Conventional Markers of Kidney Function
![]() The commonly available laboratory tests used to evaluate the patient with renal insufficiency are described in eChapter 17. Over the past 3 decades, Scr has been the most widely used laboratory test for estimating creatinine clearance (CLcr) and GFR. However, there are several limitations associated with its use. Scr varies widely with a patient’s age, gender, muscle mass, diet, and hydration status. For example, patients with reduced creatinine production, such as those with low muscle mass, may have very low Scr values (<0.6 mg/dL [<53 μmol/L]); thus, the presence of a gradual Scr rise to normal values (0.8 to 1.2 mg/dL [71 to 106 μmol/L]) may actually suggest the presence of AKI. However, in the presence of improved nutrition and a large muscle mass, a Scr of 1.2 mg/dL (106 μmol/L) may be a true representation of a person’s current renal status. Instead of using fixed numbers to determine renal function, changes in the value from a patient’s baseline need to be considered. Scr is normally inversely proportional to GFR. However, rapid changes in GFR (as they occur in AKI) disrupt this equilibrium and make Scr a very insensitive marker. In fact, changes in Scr will lag behind the GFR’s decline by 1 to 2 days due to slow accumulation, increased tubular secretion, and increased extrarenal clearance.25 This can lead to a significant overestimation of the patient’s GFR in the early stages of AKI and consequently a potential delay in the diagnosis of the syndrome.
The commonly available laboratory tests used to evaluate the patient with renal insufficiency are described in eChapter 17. Over the past 3 decades, Scr has been the most widely used laboratory test for estimating creatinine clearance (CLcr) and GFR. However, there are several limitations associated with its use. Scr varies widely with a patient’s age, gender, muscle mass, diet, and hydration status. For example, patients with reduced creatinine production, such as those with low muscle mass, may have very low Scr values (<0.6 mg/dL [<53 μmol/L]); thus, the presence of a gradual Scr rise to normal values (0.8 to 1.2 mg/dL [71 to 106 μmol/L]) may actually suggest the presence of AKI. However, in the presence of improved nutrition and a large muscle mass, a Scr of 1.2 mg/dL (106 μmol/L) may be a true representation of a person’s current renal status. Instead of using fixed numbers to determine renal function, changes in the value from a patient’s baseline need to be considered. Scr is normally inversely proportional to GFR. However, rapid changes in GFR (as they occur in AKI) disrupt this equilibrium and make Scr a very insensitive marker. In fact, changes in Scr will lag behind the GFR’s decline by 1 to 2 days due to slow accumulation, increased tubular secretion, and increased extrarenal clearance.25 This can lead to a significant overestimation of the patient’s GFR in the early stages of AKI and consequently a potential delay in the diagnosis of the syndrome.
An example of this phenomenon is illustrated by an acute renal artery thrombus that results in abrupt cessation of GFR in one kidney as a consequence of the complete obstruction of blood flow to that kidney (Fig. 28-3). Although 5 minutes following the event GFR is decreased 50% (assuming the other kidney is functioning and unaffected), the Scr remains unchanged. Assuming a standard daily creatinine production of ~20 mg/kg of lean body weight, one can expect ~1.4 g of creatinine production in a 24-hour period in a 70-kg individual. In pharmacokinetic terms, daily creatinine production is analogous to a continuous infusion, and GFR determines the elimination rate of creatinine. In a patient with normal renal function (GFR of 120 mL/min [2 mL/s]), the half-life of creatinine is 3.5 hours, with 95% of steady state achieved in ~14 hours. If GFR declines to 50%, 25%, or 10% of normal, the half-life of creatinine increases, resulting in prolongation of the time to reach 95% of steady state, specifically taking 1, 2, and 4 days, respectively.
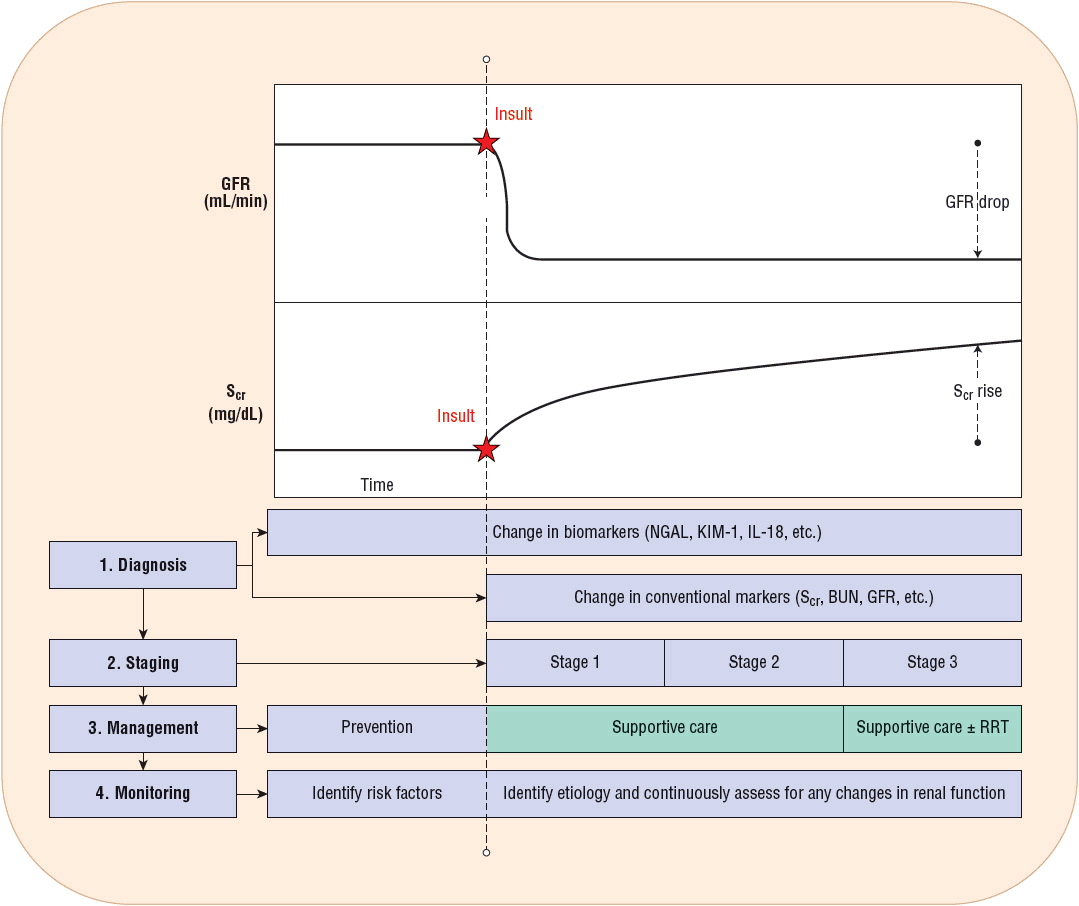
FIGURE 28-3 Glomerular filtration rate (GFR; mL/min) and serum creatinine (Scr; mg/dL) versus time following the insult that leads to acute kidney injury (AKI). Prior to the renal insult, a patient’s GFR and Scr are at stable levels. After the renal insult has occurred, GFR readily declines while Scr does not increase immediately, as it is dependent on creatinine production and attainment of steady-state serum concentrations. Novel biomarkers can detect AKI within a few hours after the injury has occurred while conventional biomarkers may take 1 to 2 days to detect a noticeable change. AKI can only be staged once Scr has increased to a significant level. Patients at risk may benefit from prevention strategies; however, once AKI is diagnosed, supportive care and potentially renal replacement therapy should be implemented. Patients with established AKI should undergo further testing to determine the most likely etiology of AKI and need to be continuously monitored for any changes in their renal function. (BUN, blood urea nitrogen; IL-18, interleukin-18; KIM-1, kidney injury molecule 1; NGAL, neutrophil gelatinase–associated lipocalin.)
Because Scr steady-state values are assumed when one uses several GFR calculation methods, such as the Cockcroft-Gault, MDRD, and Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equations, should not be used in AKI patients with unstable renal function. These equations will typically overestimate GFR when the AKI is worsening and underestimate it when the AKI is resolving. Instead, it may be useful to evaluate changes in Scr values from the patient’s baseline and also consider the Scr sequence values to determine if renal function is potentially improving (Scr values declining) or worsening (Scr values rising). The most recent Scr reflects the time-averaged kidney function over the preceding time period. Several mathematical approaches to estimate GFR in patients with unstable Scr that incorporate the principles of creatinine accumulation and elimination have been proposed and are discussed in detail in eChapter 17. However, these methods have not been extensively validated in the setting of acute alterations in renal function, and their value for adjusting medication dosing is questionable. Additionally, these equations are complex and are not commonly used in the clinical setting.
Two other widely available markers of renal function are BUN and urine output. BUN is widely used to assess hemodialysis adequacy in chronic hemodialysis patients. However, its use in AKI is very limited because urea’s production and renal clearance are heavily influenced by extrarenal factors such as critical illness, volume status, protein intake, and medications. Urine output measured over a specified period of time (e.g., 4 to 24 hours) allows for short-term assessment of kidney function, but its utility is limited to cases in which it is significantly decreased. The presence of anuria suggests complete kidney failure, whereas oliguria indicates some level of kidney damage. Urine output needs to be interpreted with caution, as it is dependent on several factors, such as hydration status and medications. As mentioned earlier in the chapter, a patient may have AKI and still maintain a normal urine output; this condition is referred to as nonoliguric AKI. Another approach to estimating renal function is to directly measure CLcr over a short period of time, for example, 4 to 12 hours.26 Although potentially precise and fairly simple to do, its accuracy is questionable if the urine output is low or the urine collection is incomplete.
In addition to BUN and Scr, selected blood tests, urinary chemistry, and urinary sediment are routinely used to differentiate the cause of AKI and guide patient management. For example, a complete blood cell count with differential can help rule out infectious causes of AKI. Serum electrolyte values may be abnormal because of the acute decline of the kidney’s ability to regulate electrolyte excretion. Particular attention should be paid to serum potassium and phosphorus values, which can be markedly elevated and cause life-threatening complications. In individuals with normal renal function, the ratio between BUN and Scr is usually less than 15:1 using conventional units (60:1 using SI units). In the presence of prerenal AKI, reabsorption of BUN exceeds that of creatinine; thus, one often sees a ratio greater than 20:1 (80:1 using SI units).
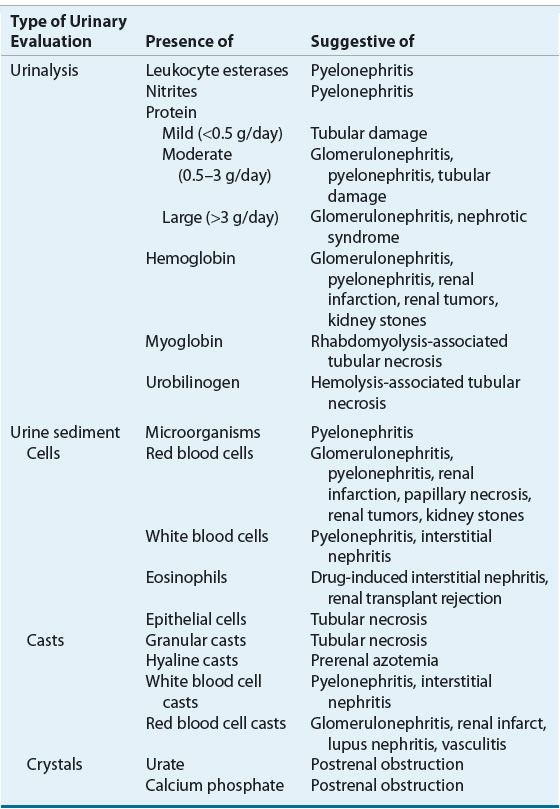
Given the limited usefulness of solely using Scr or BUN concentrations to differentiate the etiology of AKI, urinary electrolytes and osmolality should be determined, and both a microscopic and chemical analysis of the urine should be performed (Table 28-3). The finding of a high urinary specific gravity, in the absence of glucosuria or mannitol administration, suggests an intact urinary concentrating mechanism and that the cause of the patient’s AKI is likely prerenal azotemia. The presence of urinary protein is often difficult to interpret, especially in the setting of acute or chronic renal failure. A patient with CKD may have a baseline proteinuria, thus clouding the clinical presentation, unless this is known at the time of AKI assessment. Classically, proteinuria is a hallmark of glomerular damage. However, tubular damage can also result in proteinuria, as the tubules are responsible for reabsorbing small proteins that are normally filtered by all glomeruli. The presence of blood also results in a positive urine protein test, so this confounder must always be assessed when a positive urine protein is obtained. Hematuria suggests acute intrinsic AKI secondary to glomerular injury, infection, or a kidney stone. On microscopic examination, the key findings are cells, casts, and crystals, and the presence of one or more of these may suggest specific etiologies of the AKI (Table 28-4). The finding of urinary crystals may indicate nephrolithiasis and a postrenal obstruction. If red blood cells or red blood cell casts are present, one should consider the presence of a physical injury to the glomerulus, renal parenchyma, or vascular beds. The finding of white blood cells or white blood cell casts suggests interstitial inflammation (i.e., interstitial nephritis), which can be secondary to an allergic, granulomatous, or infectious process.
TABLE 28-3 Diagnostic Parameters for Differentiating Causes of AKIa
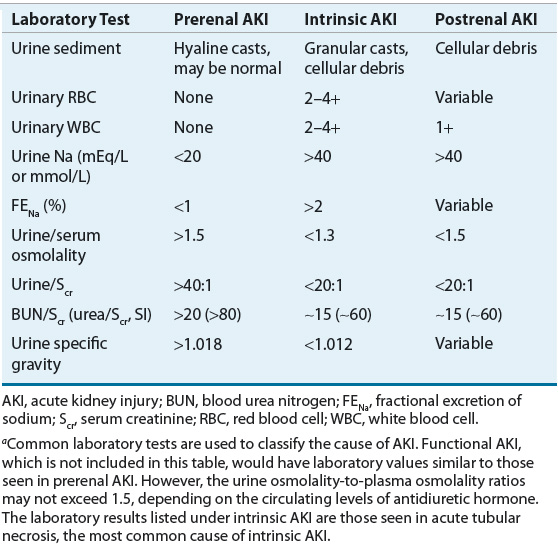
TABLE 28-4 Urinary Findings as a Guide to the Etiology of AKI
Simultaneous measurement of urine and serum electrolytes is also helpful in the setting of AKI (see Table 28-3). From these values, a fractional excretion of sodium (FENa) can be calculated. The equation for the calculation of the FENa is as follows:
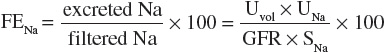
where
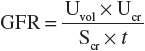
Thus:
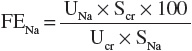
where Uvol is urine volume; Ucr is urine creatinine concentration; UNa is urine sodium; Scr is serum creatinine concentration; SNa is serum sodium concentration, which usually does not vary much; GFR is the glomerular filtration rate; and t is the time period over which the urine is collected.
The FENa is one of the better diagnostic parameters to differentiate the cause of AKI. A low urinary sodium concentration (<20 mEq/L [<20 mmol/L]) and low FENa (<1%) in a patient with oliguria suggest that there is stimulation of the sodium-retentive mechanisms in the kidney and that tubular function is intact. These findings are most characteristic of prerenal azotemia. Unfortunately, diuretic use in the preceding days limits the usefulness of the FENa calculation by increasing natriuresis, even in hypovolemic patients. The fractional excretion of urea (FEUrea), which can be calculated like FENa, is sometimes used as an alternative means to assess tubular function. The inability to concentrate urine results in a high FENa (>2%), suggesting tubular damage as the primary cause of the intrinsic AKI. However, this is also not an absolute finding, as there are some intrinsic causes that can be associated with a low FENa (e.g., contrast nephropathy, myoglobinuria, and interstitial nephritis). Highly concentrated urine (>500 mOsm/kg [>500 mmol/kg]) suggests stimulation of antidiuretic hormone and intact tubular function. These findings are consistent with prerenal azotemia.
Novel Biomarkers of Kidney Function
Diagnostic delays associated with creatinine-based methods have stimulated the search for novel biomarkers that are able to detect renal injury before a clinically evident decline in GFR occurs. Biomarkers that can detect renal injury more sensitively than Scr would enable clinicians to identify AKI earlier and, as a result, initiate preventative strategies and other interventions more rapidly.
An ideal biomarker would be highly sensitive and specific for AKI, noninvasive, and reliably and easily measurable using standardized clinical assays. In addition, it would differentiate between different AKI etiologies, be unaffected by other comorbidities and biologic variables, and allow for monitoring of response to AKI interventions. Over the past 10 years, several biomarkers have been investigated in their ability to detect and predict the clinical outcomes of AKI.27 Since no single marker fulfills all the criteria to be deemed “ideal,” combining multiple biomarkers into a panel may serve as a future clinical application for early detection, differential diagnosis, prognosis, response, and recovery of AKI. Of note, since the area of biomarker research is still relatively novel, these tests are not routinely available at most clinical practice sites. Over the past 2 years, several clinical studies evaluated the diagnostic and prognostic value of AKI biomarkers in heterogeneous patient populations and overall demonstrated promising results. However, the transition to clinical application will still require further validation, standardization, and development of implementation strategies for their use in all practice settings. Table 28-5 summarizes the advantages and disadvantages of the four most promising biomarkers.
TABLE 28-5 Advantages and Disadvantages of Novel Clinical Biomarkers of AKI
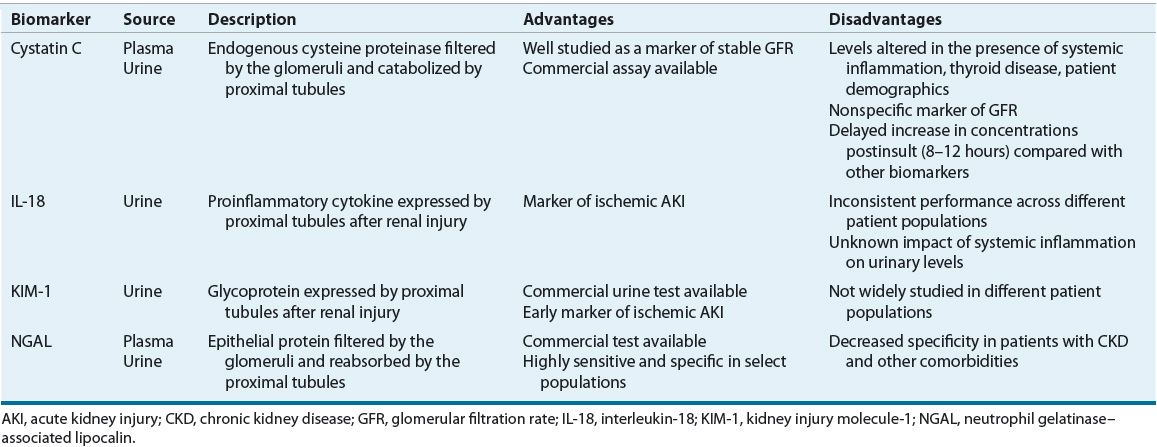
One such biomarker, serum cystatin C (see eChap. 17), is an endogenous cysteine proteinase that is released into the plasma by all nucleated cells in the body at a relatively constant rate and is then freely filtered by the glomerulus. It does not undergo any significant secretion or reabsorption, but is instead completely metabolized by the proximal renal tubules and undetectable in urine in normal kidney tissue.28 However, if tubular injury occurs, plasma cystatin C levels will rise and urinary levels will become detectable. Serum cystatin C has been extensively studied as a marker of estimated GFR in patients with stable renal function, and its measurements are readily available using standardized assays.29 Its performance in early detection and clinical outcome prediction for AKI has yielded varied results. While it does seem to outperform Scr, it has not generally outperformed other novel biomarkers.28 One of its main limitations is that, similarly to Scr, cystatin C is a marker of GFR and not a direct marker of tissue injury. Therefore, the rise in its concentrations may be delayed compared with other biomarkers and it lacks specificity in differentiating AKI from CKD.27 Also, cystatin C levels may be altered by certain disease states (e.g., thyroid dysfunction and systemic inflammation) and possibly patient demographics (age, weight, gender, etc.).30,31
Another relatively novel biomarker is neutrophil gelatinase–associated lipocalin (NGAL), a transporter protein found on cell surfaces of neutrophils and various epithelial cells. It is freely filtered by the glomeruli and reabsorbed by the proximal tubules.32 As a result, if proximal tubular injury occurs, urinary NGAL levels are expected to rise. Studies indicate that NGAL is a valuable biomarker of AKI development across a range of clinical settings, including both pediatrics and adults, patients with contrast-induced nephropathy (CIN), critically ill, and cardiac surgery patients.33 NGAL measurements may be elevated as early as 1 to 2 hours after renal injury in select populations and correlate well with the severity of AKI.34 Also, the recent development of two standardized clinical assays, a chemiluminescent microparticle assay for urine NGAL and a point-of-care kit for plasma NGAL, will help increase the availability of this measure to assess the risk of AKI.27 Although NGAL appears to be a very sensitive, specific, and early biomarker of AKI, its levels may be influenced by the presence of CKD and other comorbidities.35
Interleukin-18 (IL-18) is a proinflammatory cytokine produced by the proximal tubular epithelial cells in response to renal injury. It seems to be specific for ischemic ATN and can distinguish it from CKD, prerenal azotemia, nephrotic syndrome, and urinary tract infections.36 Urinary IL-18 levels begin increasing as early as 4 to 6 hours after an ischemic insult, peak at 12 hours, and remain elevated for up to 48 hours.37 Urinary IL-18 has been studied in several clinical settings with varied results. Its ability for early detection of AKI has proven more robust in patients with discrete ischemia reperfusion injury such as kidney transplantation or pediatric cardiopulmonary bypass and less favorable in critically ill patients or adults with comorbidities.27,38 This finding may, in part, be explained by the potential confounding impact of systemic inflammatory states on IL-18 levels, but the extent of this association still remains largely unknown.27 Even though the performance of IL-18 on early AKI detection has been inconsistent, recent studies suggest that IL-18 may be more useful as a prognostic marker of poor renal outcomes, including mortality.
Kidney injury molecule 1 (KIM-1) is a membrane glycoprotein expressed by the proximal tubular epithelial cells and released into the urine in response to ischemic renal injury. One advantage of KIM-1 is that it is not expressed in healthy kidney tissue or detected in plasma.39 While KIM-1 does have detectable levels in patients with other renal diseases such as CKD and CIN, these concentrations are significantly lower compared with ischemic AKI.27,32 This finding suggests that KIM-1 may be useful in differentiating ischemic AKI from other types of renal injury. Studies indicate that KIM-1 is a promising biomarker for early detection as its levels are elevated as early as 2 hours after renal injury.40 In addition, a rapid urine dipstick test for KIM-1 is now available that provides semiquantitative results in 15 minutes and may serve as an additional tool for rapid and early diagnosis of AKI.41
Diagnostic Considerations
When the source of renal injury is unclear after a history, physical examination, and assessment of laboratory values, imaging techniques such as abdominal radiography, including the kidneys, ureters, and bladder (KUB), computed tomography (CT), and ultrasonography may be helpful. These may reveal small, shrunken kidneys indicative of CKD. Postrenal obstruction can often be identified with a renal ultrasonogram and/or CT scan. Renal ultrasonography is also useful in detecting obstruction or hydronephrosis. Nephrolithiases as small as 5 nm or a narrowing of the ureteral tract can be detected by ultrasonography or more sensitive tests, such as KUB and CT.
In cases in which the cause of AKI is not evident, renal biopsies are useful in determining the cause in the majority of patients. Because of the associated risk of bleeding, a renal biopsy is rarely undertaken and should only be performed in those circumstances when a definitive diagnosis is needed to guide therapy, such as the precise etiology of glomerulonephritis (see Chap. 32).
PREVENTION OF AKI
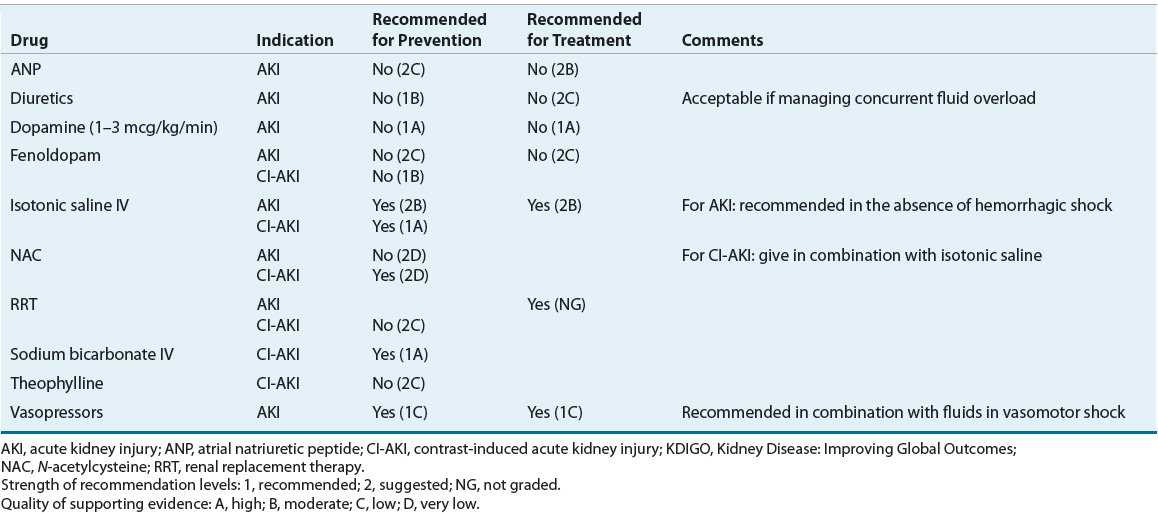
![]() The preventive strategy will depend on the type of renal insult. Clearly, complete avoidance of all potential causes of injury is the most effective preventive method; however, it may not always be possible to implement. Sometimes, the risk of renal injury is predictable, such as decreased perfusion secondary to coronary bypass surgery or secondary to the administration of a radiocontrast dye prior to a diagnostic procedure. In these situations, the potential insult to the kidneys cannot be avoided but may be preventable with aggressive hydration and removal of any additional insults. In the outpatient setting, all healthcare professionals should educate the patient on preventive measures for AKI. Patients should receive counseling regarding their optimal daily fluid intake (~2 L/day) to avoid dehydration, especially if they are to receive a potentially nephrotoxic medication. In the inpatient setting, adequate hydration, standardized hemodynamic support in the critically ill, and avoidance of nephrotoxic medications are commonly recommended strategies for the prevention of AKI. Table 28-6 summarizes the recommendations published by KDIGO clinical practice guidelines regarding recommended and not recommended therapies for the prevention of AKI.5,24
The preventive strategy will depend on the type of renal insult. Clearly, complete avoidance of all potential causes of injury is the most effective preventive method; however, it may not always be possible to implement. Sometimes, the risk of renal injury is predictable, such as decreased perfusion secondary to coronary bypass surgery or secondary to the administration of a radiocontrast dye prior to a diagnostic procedure. In these situations, the potential insult to the kidneys cannot be avoided but may be preventable with aggressive hydration and removal of any additional insults. In the outpatient setting, all healthcare professionals should educate the patient on preventive measures for AKI. Patients should receive counseling regarding their optimal daily fluid intake (~2 L/day) to avoid dehydration, especially if they are to receive a potentially nephrotoxic medication. In the inpatient setting, adequate hydration, standardized hemodynamic support in the critically ill, and avoidance of nephrotoxic medications are commonly recommended strategies for the prevention of AKI. Table 28-6 summarizes the recommendations published by KDIGO clinical practice guidelines regarding recommended and not recommended therapies for the prevention of AKI.5,24
TABLE 28-6 KDIGO Recommendations for Prevention and Treatment of AKI