Linkers
Chemical structures
Therapeutic agents
References
Acetal
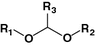
Diethylstilboestrol
[63]
Doxorubicin
[64]
Ester
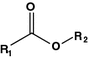
Bortezomib
[65]
Doxorubicin
[66]
Hydrazone
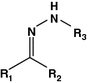
Cisplatin
[51]
Doxorubicin
Geldanamycin
[67]
Gemcitabine
[68]
Imine
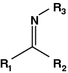
Chromone
[69]
Doxorubicin
[70]
Orthoester
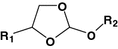
Paclitaxel
[71]
Oxime
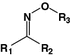
Daunorubicin
Doxorubicin
[57]
Oligonucleotide
[56]
Phosphoramidate
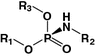
Oligonucleotide
[60]
Vinyl ether
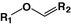
Oligonucleotide
[72]
β-Thiopropionate
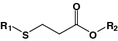
Oligonucleotide
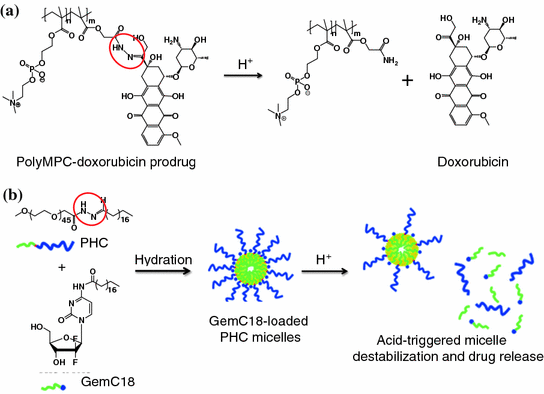
Fig. 26.1
Examples of pH-activatable agents which utilize hydrazone linker. a PolyMPC–DOX prodrugs that integrate pH-responsive hydrazone linkages into the poly(methacryloyloxyethyl phosphorylcholine) (polyMPC) structure. b Acid-sensitive micelles. The micelles were assembled by acid-sensitive copolymers, which were synthesized by conjugating the hydrophilic PEG segment to a hydrophobic stearic acid derivative with a hydrazone bond. Recreated with permission from ACS Publications [68]
In some cases, acid-sensitive linkers are incorporated into the carriers. This strategy does not require the chemical modification of a drug or an imaging agent. For example, acid-sensitive micelles have been described in the literature. They were composed of polyethylene glycol (PEG) that was conjugated to stearic acids (C18) via hydrazone linkages (Fig. 26.1b). These linkages were destabilized at a low pH and, subsequently, triggered the release of 4-(N)-stearoyl-gemcitabine (GemC18), a deoxycytidine nucleoside analog of gemcitabine used for the treatment for many cancers [68]. In another study, hydrazone linkages were introduced between the PEG chains and the phospholipids of long-circulating liposomes. Following the cleavage of the hydrazone bonding at low pH, the PEG was removed, leading to multiple cell-penetrating peptides exposing on the liposome surfaces to promote the intracellular delivery of the encapsulated doxorubicin [73].
26.2.2 Demicellization of Micelles
Some materials used to construct the acid-sensitive micelles are intrinsically sensitive to pH changes [74, 75], whereas others are introduced with functional groups that are known to be neutral at physiological pH and ionizable in an acidic environment (Fig. 26.2a). Poly(β-amino ester) is commonly used to construct acid-sensitive micelles [76, 77]. The advantage of poly(β-amino ester) is that it responds abruptly to pH changes. Given that the tertiary amino groups in the hydrophobic amino ester can be protonated (with a pKb of 6.5) in an acidic environment, the polymers can become hydrophilic. Subsequently, the micelles lose stability (dissociate) and release the encapsulated drug contents rapidly. For example, camptothecin was encapsulated in the micelles assembled from PEG and poly(β-amino ester) copolymers. The resulting micelles can be activated inside the endosomes and the lysosomes to release the drug contents [78]. Protoporphyrin IX (PpIX) is a photosensitizer that has been loaded into the pH-responsive MPEG-poly(β-amino ester) polymeric micelles [79]. The resulting micelles could be delivered to the tumors by the EPR effect and, subsequently, destabilized to release the PpIX. Upon laser irradiation, the PpIX generated near-infrared (NIR) fluorescence for tumoral imaging and, simultaneously, singlet oxygen for photodynamic therapy (Fig. 26.2b and c). In another study, to synthesize an acid-sensitive magnetic resonance imaging (MRI) probe, iron oxide (Fe3O4) nanoparticles were encapsulated inside an acid-sensitive micelle composed of copolymers of PEG, poly(β-amino ester), and poly(amido amine). This MRI probe released the Fe3O4 nanoparticles at a lower pH to image the ischemic area of the brain [80]. Other acid-sensitive micelles were also created by introducing polyhistidine or poly-4-vinylpyridine to the polymeric constructs (Fig. 26.2a) [81, 82].
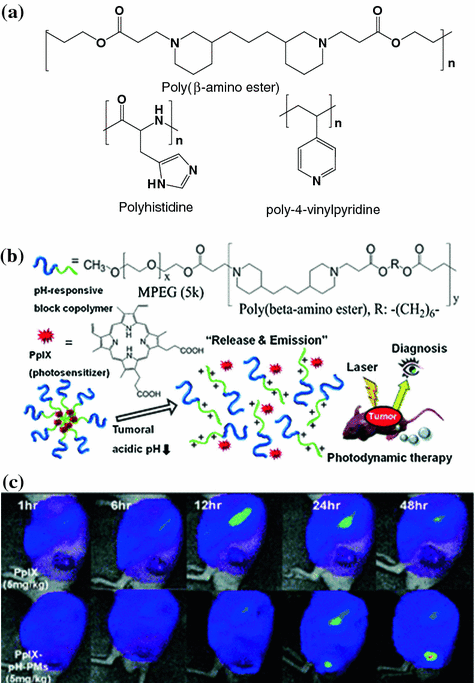
Fig. 26.2
a Examples of functional group that is neutral at a physiological pH, but ionizable in an acidic environment. b The design of the MPEG-poly(β-amino ester) micelles system. c Fluorescence images of SCC7 tumor-implanted mice injected with free PpIX (top panel) and PpIX encapsulated in the MPEG-poly(β-amino ester) micelles (PpIX-pH-PMs). Recreated with permission from RSC Publishing [79]
26.2.3 Decomposition or Desorption of Inorganic Materials
Certain inorganic materials, such as calcium phosphate (CaPO4), are insoluble in physiological pH, but can be completely dissolved at pH <6.5. Thus, pH-sensitive delivery systems have been designed either by encapsulating hydrophobic drug molecules inside the particles made up of inorganic materials [83] or by adsorbing the drugs onto the particle surfaces [84]. For instance, CaPO4 has been used to form a deposit layer on a polymer micelle through the mineralization process [85]. Doxorubicin molecules were securely trapped inside the micelle in physiological pH and were readily released after rapid dissolution of the CaPO4 layer (Fig. 26.3a). In another design, zinc oxide (ZnO) quantum dots were employed to seal the nanopores of mesoporous silica nanoparticles [86]. The dots were decomposed in an acidic environment, which caused the pores to open to promote the release of the drug contents (Fig. 26.3b).
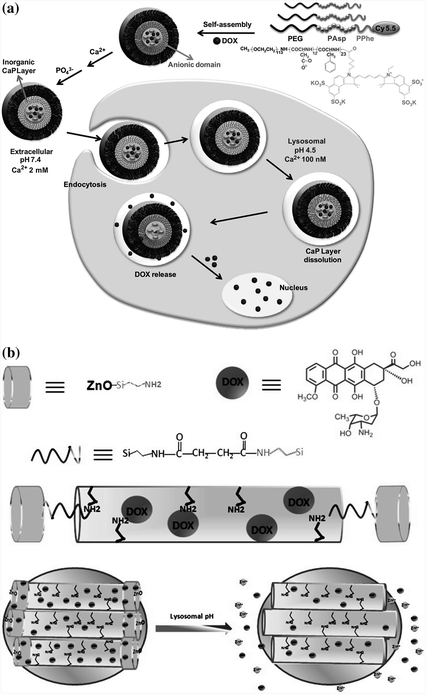
Fig. 26.3
Design of pH-sensitive agents using inorganic materials. a Calcium phosphate (CaP) was deposited onto micelles assembled from copolymers of polyethylene glycol, poly(L-aspartic acid), and poly(L-phenylalanine) through the mineralization process. The release of doxorubicin content was facilitated inside the cells at the endosomal pH because of the rapid dissolution of the CaP layer. Recreated with permission from Elsevier [85]. b ZnO quantum dots were used as a lid to seal the pores of mesoporous silica nanoparticles, which could be efficiently dissolved in the acidic intracellular compartments of cancer cells, resulting in the release of doxorubicin into the cytosol. Recreated with permission from ACS Publications [86]
Some therapeutic agents such as doxorubicin have potential metal-binding sites [87]. They can be adsorbed onto the surfaces of inorganic particles, including titanium dioxide (TiO2), magnesium oxide (MnO), and ZnO via electrostatic and hydrophobic interactions [88–92]. Despite the fact that the exact drug release mechanism is not completely understood, it can be explained as the protonation of the chemisorbed doxorubicin molecules under an acidic condition and lead to a reduction in binding to particles. Some inorganic nanoparticles, such as MnO, have been described as T1 MRI contrast agents [93]. When loaded with doxorubicin, these particles can also be theranostic agents [89, 90].
26.2.4 Microgels
Acid-sensitive microgels are composed of polymeric materials that can undergo phase transition (swelling) in response to pH [94]. At low pH, the protonation of certain functional groups, such as amino groups, generates positive charges that cause swelling. As a result, the gel permeability increases and leads to the release of drug content. Chitosan is one of the most commonly used acid-sensitive polymers for preparing microgels because it is also biocompatible. A chitosan derivative, N-[(2-hydroxy-3-trimethylammonium)propyl]chitosan chloride, has been employed to prepare acid-sensitive microgels for the delivery of methotrexate into HeLa cells [95]. The polymer has also been copolymerized with a variety of polymers, such as PEG, poly(methacrylic acid), and poly(N-isopropylacrylamide) (PNIPAM), for the delivery of other chemotherapeutic agents including 5-fluorouracil, temozolomide, and oridonin [96–98].
26.3 Enzyme-Activatable Agents
Aberration of enzyme activity or protein concentration occurs in most diseases. In the past, therapeutic agents were developed with the goal of inhibiting the action of a particular enzyme or protein. Enzyme-sensitive motifs can be incorporated into different nanoparticles, which can be readily activated by the disease-associated enzyme to release a payload at the target site [99]. The advantage of this approach is that the target enzyme behaves similarly to a self-propelled machine, where one enzyme can potentially activate hundreds to thousands of delivery systems. Further, enzyme-activatable agents can also act as substrate inhibitors. Therefore, numerous enzyme-activatable agents have been proposed for the purpose of drug delivery, therapy, and imaging in recent years [100]. To ensure the success of an enzyme-activatable agent, it is essential to have a profound understanding of the mechanism of the target enzymes (Fig. 26.4) and the heterogeneity between the disease and normal states.
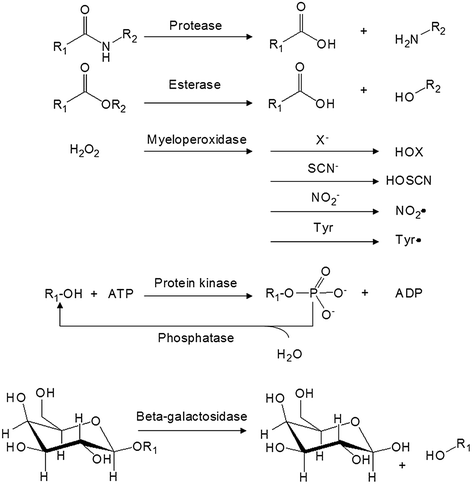
Fig. 26.4
General mechanisms of some target enzymes used to activate an enzyme-activatable agent
26.3.1 Protease
Protease belongs to a class of enzyme that has the ability to hydrolyze the amide bonding of a protein or peptide (Fig. 26.4). In 2012, there were 581 human proteases in the Degradome database, which covers approximately 2–4 % of a typical mammalian genome [101]. Each of the proteases exhibits distinct functions to maintain “normality” in a healthy state. For example, pepsin, trypsin, and chymotrypsin are secreted from the stomach and duodenum to digest proteins in food [102]. Thrombin, plasmin, and the Hageman factor are involved in the normal function of blood clotting [103]. Inside a cell, some proteases, such as cathepsin D, are stored within the lysosomes for breaking down waste material and cellular debris. Other intracellular proteases are known to regulate gene expression, cell growth, and apoptosis [104, 105]. Because of the diverse functionality and abundant amount of proteases, many of them are abolished or elevated during disease progression. For instance, dysregulated protease activities are commonly found in atherosclerosis [106–108], thrombosis [109], arthritis [110], asthma [111], emphysema [112], and osteoporosis [113]. In the context of cancer, cathepsins [114], matrix metalloproteinases (MMPs) [115], urokinase plasminogen activators (uPA) [116], and prostate-specific antigen (PSA) have been shown to play distinct roles in tumor growth, invasion, migration, and angiogenesis by facilitating the breakdown of extracellular matrix (ECM) [116–118].
Advances in solid-phase peptide synthesis and combinatorial chemistry have led to the discovery of many peptide sequences that are specific to individual proteases [119, 120]. When these peptides are conjugated to chemotherapeutic agents, the resulting conjugates become prodrugs that can be activated by targeted proteases [121–127]. A peptide (Mu-HSSKLQL) that is specific to PSA cleavage was coupled to the primary amine of doxorubicin. The drug–peptide conjugate exhibited a specific cytotoxic response to PSA-positive prostate cancer cell lines [128]. In another example, a synthetic semenogelin I peptide analog was covalently conjugated to the aminoglycoside of doxorubicin. The resulting L-377,202 showed a significant reduction in dose-limiting systemic toxicities compared to free doxorubicin [129]. In the clinical context, L-377,202 was well tolerated in patients with advanced hormone-refractory prostate cancer. The prodrug could be cleaved by PSA to release the active metabolites [130]. The similar design has been applied to numerous other drug–peptide conjugates (Table 26.2). Unfortunately, peptide-based therapeutic agents normally exhibit short elimination half-lives. Further, they are difficult to administer over a prolonged dosing interval [131].
Table 26.2
Some of the drug–peptide conjugates
Active drug metabolite | Peptide substrate | Target protease | Disease | References |
|---|---|---|---|---|
Amiloride | LFGGGG | Enkephalinase | Ischemia | [136] |
Cyclopamine | SSKYQ | PSA | Cancer | [137] |
Cyclophosphamide | Cbz-SSFY | PSA | Cancer | [138] |
Doxorubicin | BLAL | TOP | Cancer | [139] |
PLGL, PVGLIG | MMP-2/9 | Cancer | ||
aFK | Plasmin/urokinase | Cancer | ||
RSSYYSR | PSA | Cancer | [144] | |
AANL | Legumain | Cancer | [145] | |
Fluorodeoxyuridine | HSSKLQL | PSA | Cancer | [146] |
Gentamicin | GfPRGFPAGG | Thrombin | Infection | [147] |
Methotrexate | aFKK | Cathepsin, plasmin | Arthritis | [148] |
Paclitaxel | aFK, vLK | Plasmin | Cancer | [149] |
V-Cit | Cathepsin B | Cancer | [150] | |
GPLGIAGQ | MMP-2 | Cancer | [151] | |
Pyropheophorbide-a | TSGPNQEQK | FAP | Cancer | [152] |
DEVD | Caspase 3 | Cancer | [153] | |
TGX-221 | SSKYQ | PSA | Cancer | [154] |
Thapsigargin | HSSKLQL | PSA | Cancer | [155] |
GKAFRR | Hk2 | Cancer | [156] | |
Vinblastine | Hyp-SS-Chg-QSSP | PSA | Cancer | [127] |
To increase circulation time, these prodrugs can be covalently attached to a biomolecule. Antibodies are frequently used as the biomolecules because they are specific. These so-called antibody–drug conjugates can deliver the therapeutic agents inside the target cell. In a successful case, monomethylauristatin E (MMAE) was conjugated to a monoclonal antibody (cAC10) that was specific CD30 via a dipeptide (V-Cit) [132]. In vitro study showed that the resulting antibody–drug conjugate (brentuximab vedotin) could target CD30-expressing malignant cells and could be cleaved by cathepsin B to release the active MMAE. In vivo study showed that brentuximab vedotin was able to suppress subcutaneous Karpas 299 anaplastic large cell lymphoma (ALCL) tumors [132]. In 2011, brentuximab vedotin was approved by the FDA for the treatment for ALCL and Hodgkin’s lymphoma [133]. However, some studies have shown that using peptides might not be superior to using a pH-cleavable linker when designing an antibody–drug conjugate [134]. Occasionally, a protein may be selected as the biomolecular carrier. For example, paclitaxel has been conjugated to albumin via a peptide (RSSYYSL) that is specific to PSA cleavage. The albumin improved the pharmacokinetic and the solubility of the drug. The macromolecular prodrug was three times more potent toward PSA-positive LNCaP prostate cancer cells, and the maximum-tolerated dose in vivo was twice that of free paclitaxel [135].
A number of macromolecular prodrugs have been designed using synthetic polymers (Table 26.3). N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers have been used extensively. For example, prostaglandin E1 (PGE1) has been conjugated to HPMA through a cathepsin K-sensitive peptide linker (GGP-Nle, where Nle = norleucine) [157]. Incubation of the polymer with cathepsin K-expressing osteoblasts caused a rapid release of PGE1 for the treatment for osteoporosis [158]. The same polymer has been used to deliver many other chemotherapeutic agents including 5-fluorouracil, anthracycline, platinum (II), and TNP-470 for the treatment for different cancers [159–164], except the drugs were conjugated to the polymer via a cathepsin B-sensitive peptide (GFLG).
Table 26.3
Some of the macromolecular prodrugs
Macromolecule | Active drug metabolite | Peptide substrate | Target protease | Disease | References |
|---|---|---|---|---|---|
Protein | |||||
Avidin | Equinatoxin II | CNKSRLGLGK | Cathepsin B/MMP-7 | Cancer | [165] |
Albumin | Camptothecin | RRALALA | Cathepsin B | Cancer | [166] |
Doxorubicin | FK, RRALAL | Cathepsin B | Cancer | ||
GPLGIAGQ | MMP-2/9 | Cancer | |||
RSSYYSR | PSA | Cancer | [144] | ||
GGGRR | Urokinase | Cancer | [170] | ||
Paclitaxel | RSSYYSL | PSA | Cancer | [135] | |
Antibody | |||||
CBR96 | MMAE | V-Cit | Cathepsin B | Cancer | [132] |
cAC10 | MMAE | V-Cit | Cathepsin B | Lymphoma | [132] |
Polymer | |||||
CM-Dextran-PA | Exatecan | GGFG | Cathepsin B | Cancer | [171] |
Dextran | Methotrexate | PVGLIG | MMP-2/9 | Cancer | [172] |
HPMA | 5-Fluorouracil | GFLG | Cathepsin B | Cancer | [159] |
Doxorubicin | GFLG | Cathepsin B | Cancer | ||
Daunorubicin | GFLG | Cathepsin B | Leukemia | [162] | |
L12ADT | SSKYQ | PSA | Cancer | [173] | |
Platinum (II) | GFLG | Cathepsin B | Cancer | [163] | |
Prostaglandin E1 | GGP-Nle | Cathepsin K | Osteoporosis | ||
TNP-470 | GFLG | Cathepsin B | Cancer | [164] | |
PGA | Paclitaxel | (E)x | Cathepsin B | Cancer | [174] |
Recently, many fluorophores, including fluorescein, indocyanine green (ICG), cresyl violet, isosulfan blue, methylene blue, and porphyrins, have been identified as contrast agents for optical imaging [175–181]. By simply replacing the drug molecule with a fluorophore to conjugate it to a macromolecule, a protease-activatable probe can be developed (Fig. 26.5). For example, multiple peptides (GSGRSANA) capped with different NIR fluorophores (Cy5.5 or Cy7) have been conjugated to PEG and poly-L-lysine copolymer. The probe was optically silent (quenched) in its native state, as the fluorophores orientated in a close proximity. Upon digestion by a serine protease (uPA), the polymer released fluorophore–peptide fragments, which resulted in the recovery of fluorescence [182]. The probe was used to visualize the uPA activity in HT1080 tumor xenografts [183]. Using the same strategy, other peptide substrates have also been applied to imaging other disease-associated proteases including cathepsins, MMPs, caspases, and thrombin [184–189].
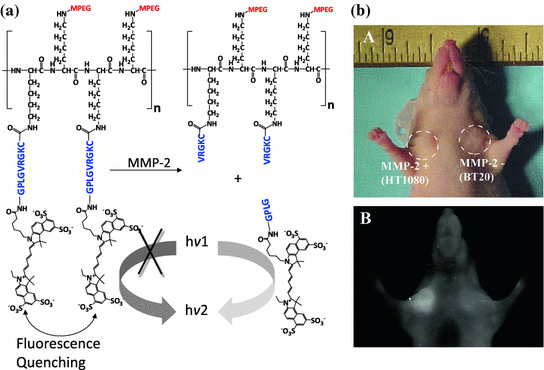
Fig. 26.5
An enzyme-activatable probe designed to be activated by MMP-2-associated protease. a The probe was composed of multiple peptide motifs that were terminally capped with different Cy5.5 and a pegylated poly-L-lysine graft copolymer. b In vivo imaging. NIRF imaging of a nude mouse implanted with HT1080 (MMP-2+) and BT20 (MMP-2−) tumors. Photograph and fluorescence image of the mouse 2 h after intravenous injection of the probe. Recreated with permission from RSNA [187]
For therapeutic agents that are too fragile to be chemically modified or do not have a spare functional group for bioconjugation, a drug can be encapsulated in a carrier that can be degraded by proteases. Some materials used for engineering nanoparticles, such as gelatin, can be intrinsically degraded by proteases; therefore, they have been used for the delivery of chemotherapeutic agents, such as paclitaxel [190]. On the other hand, enzyme-activatable nanoparticles have been custom-designed by incorporating peptide substrates as part of the components. Short chains of PEG (~480 Da) have been cross-linked with peptide substrates (RVRR) to form nanocapsules to protect and carry a recombinant myogenic transcription factor across the cell membrane [191]. Upon uptake by the cell, the intracellular furin, a ubiquitous endoprotease, degraded the capsules and, subsequently, released the encapsulated protein. In another example, sodium poly(styrene-4-sulfonate), a polymeric microbicide used to treat HIV infection, was encapsulated in the microgel particles composed of HPMA and 2-aminopropylmethacrylamide (APMA) copolymers cross-linked with PSA substrates (GISSFYSSK) [192]. The resulting particles were degraded by the PSA in human seminal plasma (HSP) and released the entrapped antiviral polymer.
26.3.2 Esterase
Esterase is an enzyme that breaks down the ester, amide, and thioester bonding of an organic molecule. It can be found in many organs including the liver, lung epithelia, small intestine, kidney, and plasma [193]. To improve the solubility and/or bioavailability of the drug formulation [194–198], prodrugs such as capecitabine, oseltamivir, meperidine, and methylphenidate were designed. They can be converted by carboxylesterase to the active pharmacophores (Fig. 26.6). Esterase-activatable nanoparticles have only been reported recently. Lipid nanoparticles assembled from ester-containing polysorbate 60 and PEG6000 monostearate have been synthesized [199]. Silica mesoporous nanoparticles (MCM-41) have been employed for the delivery of camptothecin [200]. The pore outlets of MCM-41 were capped with ester glycol moieties as the stoppers, which could be hydrolyzed by esterase in the lysosome inside HeLa cells to release the active drug contents. In another example, micelles were constructed from amphiphilic dendrimers functionalized with hexyl ester (as the lipophilic unit) and pentaethylene glycol (as the hydrophilic unit). In the presence of porcine liver esterase, the enzyme hydrolyzed the ester functionalities and caused the demicellization of the micelles, leading to the release of hydrophobic guest molecules at the target site [201].
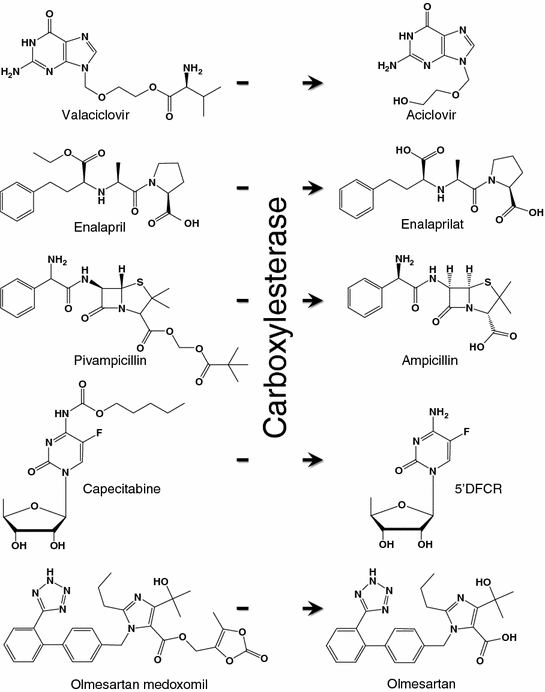
Fig. 26.6
Examples of carboxylesterase-activatable prodrugs and their metabolites
26.3.3 Myeloperoxidase
Myeloperoxidase (MPO) is an enzyme that is predominantly stored in azurophilic granules of polymorphonuclear neutrophils and macrophages. The enzyme consumes hydrogen peroxide to produce hypochlorous acid and tyrosyl radicals during neutrophil’s respiratory burst. During inflammation, MPO is released into extracellular fluid; consequently, it has been implicated in many diseases including Alzheimer’s disease, atherosclerosis, Parkinson’s disease, stroke, and vasculitis [202–206]. For these reasons, a number of myeloperoxidase-activatable contrast agents have been developed for MRI [207–210]. They were designed with the attachment of an MPO substrate, such as 5-hydroxytryptamine-(serotonin) and tyramide, to a paramagnetic gadolinium chelate, such as DOTA-Gd or DPTA-Gd, via amide bonding (Fig. 26.7). Upon enzyme activation, these contrast agents form radical species and oligomerize or bind to proteins, which results in an increase in the T1-weighted signal in MRI. For example, bis-5HT-DTPA-Gd has been used to image the MPO activity of a living animal with cerebral ischemia [211]. The post-contrast T1-weighted image of the brain was enhanced compared to the conventional DTPA-Gd (Fig. 26.7). The same contrast agent has also been applied to visualize the MPO activity in atherosclerotic plagues and myositis [212, 213].
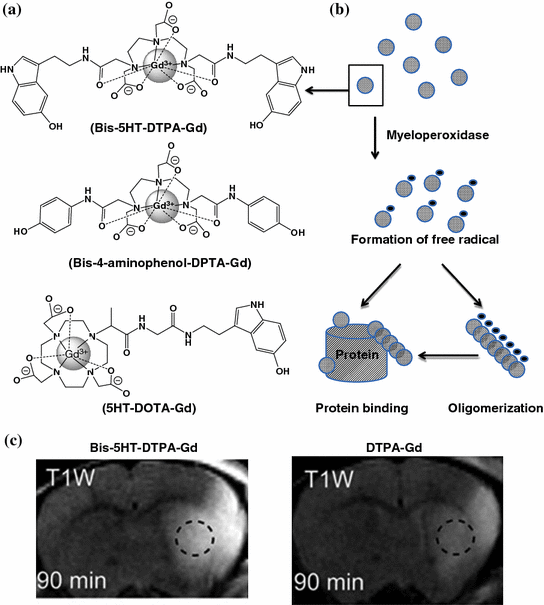
Fig. 26.7
Myeloperoxidase-activatable MRI contrast agents. a The chemical structures of some of the developed myeloperoxidase-activatable contrast agents. b Upon enzyme activation, bis-5HT-DTPA-Gd radicalized to form oligomers and/or to bind to other proteins, which resulted in the increase in r1 relaxivity. c A comparison of the T1-weighted brain images of animals injected with bis-5HT-DTPA-Gd and DTPA-Gd. Recreated with permission from PNAS [211]
26.3.4 Miscellaneous Enzymes
Numerous innovative agents have been for activation by other enzymes. However, their application in vivo still needs to be addressed. A multiple protein kinase A (PKA) substrate kemptide, and an NIR fluorophore, Cy5.5, have been incorporated into positively charged polyethylenimine [214]. The polymer was coassembled with the negatively charged polyacrylic acid (PAA) to form an optically silent polyion-induced complex (PIC). The PIC has been used for imaging protein phosphorylation in single living cell. Upon enzyme activation, the particle became phosphorylated, dissolved, and resulted in the recovery of NIR fluorescence. The same peptide has also been grafted to polyacrylamide, resulting in the formation of a cationic polymer (PAK) [215]. PAK formed a stable complex with DNA and halted gene expression. Ex vivo study showed that the activated protein kinase A (PKA) in abnormal cells could destabilize the complex via phosphorylation to release the DNA for gene transcription. Other prodrugs that can be activated by β-galactosidase were also developed as potential candidates for chemotherapy [216–220] and imaging purposes [221].
26.4 Thermo-Activatable Agents
In certain medical conditions such as hyperpyrexia, body temperature can be elevated up to 41.5 °C. In other disease states, including cancer, the microenvironment of tumors is slightly hyperthermic. In the past, numerous thermo-activatable agents have been developed for drug delivery with the aim of enhancing local drug release and cellular uptake. Another advantage of applying additional heat [i.e., hyperthermia therapy (HT)] to the tumor is that it can sensitize the cancer cells when responding to radiation and chemotherapy; therefore, HT has been proposed as an adjunctive therapy [222–226]. Many micelles have been assembled from thermo-responsive polymers (Table 26.4). In particular, PNIPAM has been widely used as the thermo-activatable polymer. It has a lower critical solution temperature (LCST) of 32 °C and can be dissolved at temperature below 32 °C. When the temperature is above the LCST, the polymer can undergo phase transition to form a gel. Thus, PNIPAM has been used in combination with other hydrophobic constructs. The resulting copolymers were able to assemble into a micelle consisting a thermo-responsive coating (Fig. 26.8). Furthermore, therapeutic agents, such as doxorubicin, could be encapsulated inside micelles. Upon heating, the coatings collapsed and resulted in the release of drug contents and/or enhanced cell attachment [227, 228]. Docetaxel has been loaded into micelles assembled from copolymer, poly(N-isopropylacrylamide-co-acrylamide)-b-poly(DL-lactide). The cytotoxicity of the micelles was significantly increased with hyperthermia therapy (Fig. 26.9) [229]. In vivo study using human BCG gastric tumor-implanted nude mice demonstrated that HT could significantly enhance the tumor inhibition of the docetaxel-loaded micelles compared to animals without hyperthermia treatment (82 vs. 32 %) (Fig. 26.9) [230]. Other newer types of polymers have also been used to generate thermo-activatable particles or micelles [231–233]. However, their performance still needs to be confirmed in vivo.
Table 26.4
Some of the polymers employed to assemble into thermo-activatable micelles
Polymers | Drug | LCST (°C) | References |
|---|---|---|---|
PIPROX-PAA | N/A | 32 | [231] |
PNIPAM-PBMA | Doxorubicin | 33 | [234] |
PNIPAM-(PHEMA –PCL)x | Paclitaxel | 36 | [235] |
(PNIPAM-PNHMAM)x-PLLA | Methotrexate | 37 | [236] |
(PNIPAM-PHEMA)x-(PLLA-PCL)y | Doxorubicin | 38 | [237]
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|