CHAPTER OUTLINE
5.2 Three Theories of Acids and Bases
5.3 Self-Ionization of Water and the pH Scale
5.4 Avoiding Confusion—Use of “Acid” and “Base” and Related Terms
5.5 The Acid Dissociation Constant Ka and pKa as a Measure of Acid Strength
5.6 Electronegativity and Size of Atoms and Acid/Base Strength
5.7 Atom Hybridization and Acid/Base Strength
5.8 Resonance Electronic Effects on Acid/Base Strength
5.9 Inductive Electronic Effects of Substituents on Acid/Base Strength
5.10 Combined Inductive and Resonance Effects on Acid/Base Strength
5.11 Proximity and Through-Space Effects on Acid/Base Strength
5.12 The Henderson–Hasselbalch Relationship and Acid/Base Equilibria as a Function of pH
5.14 Case Study—Discovery of Tagamet
5.1 Introduction
While carbon (C) and hydrogen (H) form the foundation of organic molecules, the rich diversity and specificity of interactions between biological and drug molecules arises from the presence of heteroatoms (N, O, S, P, halides) when they combine with C and H to form various functional groups. One property of many heteroatom containing molecules is a certain acidity or basicity at physiological conditions that can contribute to the reactivity and physiochemical properties of the molecule. Although drug molecules are often designed to have little or no chemical reactivity as administered, once absorbed they are metabolized (i.e., undergo chemical conversions catalyzed by the enzymes of drug metabolism) to give products with new functional groups having enhanced reactivity, potentially leading to adverse side effects. In addition to influencing reactivity, acid/base properties and the charge state of a drug molecule contribute significantly to its relative solubility in water (as found in the interior of cells and in bodily fluids) versus nonpolar media (as found in the lipid membranes of cells). This differential solubility impacts the absorption of the drug and hence the amount that needs to be administered to achieve the desired effect. In this chapter, we review the principal models of acid and base behavior and discuss the relationship of structure and bonding to the acidity of different types of “X–H” bonds (C–H, N–H, O–H, S–H) and the basicity of the lone pairs of electrons on the corresponding “X” atoms (:N, :O, :S).
5.2 Three Theories of Acids and Bases
In the late 1800s, Arrhenius proposed a theory of acids and bases based on observations of “what happens” when a substance is dissolved in pure water. The key observation was that some substances cause an increase in the hydrogen ion concentration, [H+], when dissolved in water and others cause an increase in the hydroxide concentration, [OH−]. On this basis, Arrhenius defined an acid as a compound that dissociates in water to give H+ and an anion (e.g., HCl → H+ + Cl−), and a base as a compound that dissociates in water to give a cation and OH− (e.g., KOH → K+ + OH−). Although the noted changes in [H+] and [OH−] can be a useful description of acid/base behavior for certain systems, the model is limited in the scope of acids/bases and solvents (water only) that can be described.
A more general theory of acids and bases that focuses specifically on “who has the proton” was proposed separately in 1923 by Brønsted in Denmark and by Lowry in England. In this model, the proton never exists in solution as an isolated ion because it is energetically too unstable. Instead it is always covalently bonded to another atom, but can be transferred between lone pairs of electrons on two different atoms. In this proton transfer reaction, the proton donor is the Brønsted–Lowry acid and the proton acceptor, that is, the atom (or atom within a molecule) that takes the proton from the acid, is the Brønsted–Lowry base (Figure 5.1). The Brønsted–Lowry model of acids and bases is the foundation for our discussions in this chapter.

Figure 5.1 Proton transfer reaction between a Brønsted–Lowry acid and base.
In a typical Brønsted–Lowry acid-base reaction (Figure 5.1) the initial acid (HA) is converted to its conjugate base (:A−), and the initial base (B:) is concomitantly converted to its conjugate acid (BH+). Significantly, this approach to acid/base chemistry focuses attention on the molecular structures of the two bases that transfer H+ between them (B: and :A− in Figure 5.1) allowing a direct comparison of the relationship of structure to basicity. Furthermore, since the acid and base forms of the same molecule (e.g., H–A and :A−) differ by only a single proton (H+), analysis of trends in acidity or basicity as a function of molecular structure provides significant insight into the relationship of structure to reactivity.
The Brønsted–Lowry definition is much broader in scope and works for different solvents, since H+ can be transferred between bases in any solvent. However, the behavior that Arrhenius observed for bases and acids in water is also consistent with the Brønsted–Lowry definition, in the sense that water (H2O) can act as either a base to accept a proton from another acid or as an acid to donate a proton to a base (Figure 5.2). Molecules like water that can act as both an acid and a base are called amphoteric molecules. Conceptually, the only difference between the models for the reaction of a neutral HA acid in water is that the proton is transferred to a water molecule to produce a hydronium ion rather than simply dissociating to H+. Note that only one of the two lone pairs on oxygen is shown in the acid-base reaction involving water (Figure 5.2). For simplicity’s sake, we will generally show only the lone pair involved in the acid-base reaction in the schemes and tables of this chapter.

Figure 5.2 Reactions showing the relationship between the Brønsted–Lowry and Arrhenius definitions of acids and bases. Only those lone pair electrons involved in the acid-base reaction are shown explicitly.
A completely separate notion of acids and bases was devised by G. N. Lewis in 1923 and formally proposed in 1938. Rather than focusing only on protons, Lewis’ theory asks “who has the electrons?”. Thus, a Lewis acid is an electron acceptor and a Lewis base is an electron donor. With this definition, any atom that has a positive charge, a partial positive charge, or an unfilled valence shell of electrons is considered a Lewis acid. Metal ions such as Zn2+, Ca2+, and Mg2+ are common positively charged Lewis acids of importance in biological systems. Carbons found in highly polar bonds such as in a carbonyl group (δ+ C=O δ−) are important Lewis acids involved in many important biological reactions. Since boron has only three valence shell electrons of its own, compounds like BF3 are two electrons short of an octet, and thus are very strong Lewis acids (if not biologically relevant ones). Essentially any Brønsted base can also be considered a Lewis base since both utilize a lone pair of electrons to react with their corresponding acids (Figure 5.3).

Figure 5.3 Reaction illustrating Lewis theory definition of acids and bases.
Note that whereas a Brønsted acid gives up its proton to a different Brønsted base when they react, a Lewis acid combines (i.e., forms a bond) with a Lewis base when they react (Figure 5.3). The Lewis conception of acids and bases is very useful for discussions of chemical reactivity and we will return to it in Chapters 6 and 7.
5.3 Self-Ionization of Water and the pH Scale
The observation that pure water has low levels of H+ and OH− is easily understood by the Brønsted–Lowry theory. In this self-ionization behavior, one molecule of water acts as the proton donor (acid) and a second molecule acts as the proton acceptor (base) to generate hydroxide and a hydronium ion (H3O+).

As noted in the equation above, thermodynamic equilibrium constants (Keq) are formally defined by the activities (ai) of each species. However, for dilute solutions where the solvent is in vast excess, the activity of the solvent (water in this case) is unity (aH2O = 1) and the activities for the dilute ions are essentially equal to their molar concentrations (aH3O+ ~ [H3O+], aOH− ~ [:OH−]). As a result, the equilibrium dissociation constant for the self-ionization reaction of water (its acid dissociation constant) is simplified to
Kw = [H3O+][: OH−]
At 25°C, Kw = 10−14 M2, and since the self-ionization reaction yields equal amounts of H3O+ and OH−, both are present at 10−7 M in pure water. As other acids or bases are dissolved in and react with water, the concentrations of H3O+ and OH− re-equilibrate such that their product always equals 10−14. Thus when [H3O+] = 1 M, [OH−] = 10−14 M and vice versa. To simplify discussions of the relative acidity of aqueous solutions, we use the pH scale, where
pH = – log10[H3O+]
For pure water with [H3O+] = 10−7 M, the pH = 7, which is also referred to as neutral pH because the concentrations of H3O+ and OH− ions are equal. Acidic solutions have higher concentrations of H3O+ ([H3O+] > [OH−]) and pH values < 7, whereas basic solutions have lower concentrations of H3O+ ([H3O+] < [OH−]) and pH values > 7.
5.4 Avoiding Confusion—Use of “Acid” and “Base” and Related Terms
In our descriptions of the three theories of acids and bases above, we found that the terms “acid” and “base” can mean several different things depending on the context in which they are used. An acid can be (1) a substance that increases the concentration of protons (hydronium ions), [H3O+], when dissolved in pure water—Arrhenius acid; (2) a proton donor—Brønsted–Lowry acid; (3) an electron pair acceptor—Lewis acid. Conversely, a base can be (1) a substance that increases the concentration of hydroxide ions, [OH−], when dissolved in pure water—Arrhenius base; (2) a proton acceptor—Brønsted base; (3) an electron pair donor—Lewis base.
Two additional pairs of terms used in discussions of acids and bases, acidic/basic and acidity/basicity, may also bring confusion as they are used in more than one context. The most common contexts for each pair include:
The terms “acidic” and “basic” are typically used in four contexts. The first is to describe the property of a solution. Any solution where [acid] > [base] could be described as “acidic,” and the converse situation where [base] > [acid] could be described as “basic.” Aqueous solutions of pH lower than 7, where [H3O+] > [OH−], are acidic and those of pH higher than 7, where [OH−] > [H3O+], are basic. The second context is to describe the property of a compound. For example, any acid can also be called an acidic molecule or an acidic compound, and conversely, any base can also be called a basic molecule or a basic compound. The third context is to describe a property of a functional group. Functional groups that can act as a proton donor are acidic functional groups, while those that can act as a proton acceptor are basic functional groups. For example, the carboxylic acid group, which has the general formula RCOOH, is an acidic functional group, and an amine, which has the general formula R3N: is a basic functional group. Finally, the fourth context is to describe the property of a specific X–H bond in a Brønsted–Lowry acid or a specific lone pair of electrons in a Brønsted–Lowry base. Using the same examples, the specific bond that is acidic in a carboxylic acid is the O–H bond and the specific pair of electrons that is basic in an amine is the N: lone pair.
The terms “acidity” and “basicity” are typically used in two contexts. The first is to describe how far the pH of an aqueous solution deviates from pH 7. The acidity of a solution varies from weak to strong as the pH decreases from 7 to 0, while the basicity of a solution varies from weak to strong as the pH increases from 7 to 14. The second context is to describe the relative strength of an acid or base, respectively. The more readily a Brønsted–Lowry acid gives up its proton, the greater is its acidity. The more attracted a Brønsted–Lowry base is to a proton, the stronger is its basicity.
5.5 The Acid Dissociation Constant Ka and pKa as a Measure of Acid Strength
The relative strengths of acids are determined by comparisons of their acid dissociation constants. Although these have been measured in different solvents, the most commonly used and most relevant acid dissociation constants for discussing molecules in biological systems are those measured in water with concentrations expressed in units of moles per liter (M).

Although water is a reactant (Brønsted base) in this equilibrium, its concentration does not appear in the equation for the acid dissociation constant (Ka) for the same reason noted above for the self-ionization of water. Since the acid and base forms of a molecule (e.g., H–A and :A−) only differ by a single proton (H+), any structural features that make H–A a stronger acid must also make its conjugate base :A− a weaker base. Thus, we can evaluate trends in the strengths of acids and bases by considering how structural features affect either the acidity of H–A or the basicity of :A−. For this reason, and to simplify comparisons of various kinds of acids and bases, we typically evaluate the strength of uncharged bases B: by determining an acid dissociation constant (Ka) for the conjugate acid BH+ as it transfers its proton to water (as illustrated in the following equation). In this way both the acidity of H–A acids and the basicitiy of B: bases can be compared on the same scale (Ka or pKa as defined below).

Very strong acids such as HI, HBr, and H2SO4 dissociate almost completely when dissolved in water, so their Ka values are all  1 M. (These cannot actually be measured in water but can be determined relative to water using other solvents.) On the other hand, functional groups in organic molecules are typically weak or very weak acids and have Ka values ranging from 10−1 down to 10−50 M. To simplify discussions of this large range of values, acid dissociation constants are expressed as pKa (also called acidity constants) where
1 M. (These cannot actually be measured in water but can be determined relative to water using other solvents.) On the other hand, functional groups in organic molecules are typically weak or very weak acids and have Ka values ranging from 10−1 down to 10−50 M. To simplify discussions of this large range of values, acid dissociation constants are expressed as pKa (also called acidity constants) where
pKa = – logKa
This is analogous to the use of pH to express hydronium ion concentrations, as discussed already. Using this scale, acids with a pKa < 0 are very strong acids and the acidity of X–H bonds decreases as the pKa increases above 0. Very weak acids with conjugate bases stronger than hydroxide ion do not dissociate to any significant extent in water and have Ka values < Kw = 10−14 (pKa > 14). (These also cannot be measured in water but are estimated relative to water using other solvents.)
Tables 5.1 and 5.2 list structures of acid and conjugate base forms of the most common HA and BH+ type functional groups found in biological and drug-like molecules along with their pKa values. The structures shown in the table have only a single functional group to underline the relationship of the specific structural features in these molecules to their relative acidity (and basicity). The acidic proton is colored blue in each example and the structural features contributing to the basicity of the X: atom (and corresponding acidity of the X–H bond) are colored red and range from a single atom in some cases to the whole molecule in others. The tables are organized by descending pKa (very weakly to very strongly acidic) from top to bottom. The trend in the strength of the corresponding conjugate base is of course in the opposite direction, from very strong bases at the top to very weak ones at the bottom. Thus, when comparing two functional groups, the one with a higher pKa value is the weaker acid and has the stronger conjugate base. We will refer back to these tables in later sections as we discuss how structural features of molecules and their substituents affect acidity and basicity.
Table 5.1 Acidity Constants (pKa) of Selected HA Type Acids: C–H, N–H, O–H, S–H, H–X.



Table 5.2 Acidity Constants (pKa) of Selected BH+ Type Acids: +N–H, +O–H, +S–H.



5.6 Electronegativity and Size of Atoms and Acid/Base Strength
Two intrinsic properties of atoms affect the strengths of X–H bonds independent of other structural features. The first of these is the electronegativity of X, which was introduced in Chapter 1 and can be compared for elements within the same period (i.e., row of the periodic table) that use the same valence shell electrons to form bonds (Table 5.3).
Table 5.3 Electronegativities of Elements in the First Three Rows of the Periodic Table.

The effect of electronegativity on acidity can be seen in the acidities (pKa values) of the simple compounds CH4, NH3, H2O, and HF, based on elements from the second row of the periodic table (Figure 5.4).

Figure 5.4 Example of effect of electronegativity on the acidity of X–H bonds in similar molecular contexts. (Reproduced, with permission, from Carey FA, Giuliano RM. Organic Chemistry. 9th ed. New York: McGraw-Hill Education; 2014.)
As the electronegativity of the X: atom increases in this series, the polarity of the X–H bond increases so that the H carries a larger partial positive charge (δ− X–H δ+) and the equilibrium for proton transfer to water increases (the acid becomes stronger). Another way to look at this series is that as the X atom becomes more electronegative, it can stabilize negative charge better and thus let go of a proton more easily. With regard to the conjugate base :X−, we would say that the more electronegative the atom X, the weaker the base :X−. So comparing across rows, increasing electronegativity of the X atom increases acidity of X–H and decreases basicity of :X−.
The trend also holds for acids of the BH+ type as seen in the following example where the more electronegative oxygen is much less basic than nitrogen in a similar context (Figure 5.5).

Figure 5.5 Effects of electronegativity on the acidity of +X–H bonds in similar molecular contexts (elements in the same row of the periodic table).
The negative pKa value indicates that protonated methanol (CH3OH2+) is quite a strong acid and therefore that neutral O: in methanol (CH3OH) must be a very weak base. By contrast, the pKa of methyl ammonium ion (CH3NH3+) indicates that it is a very weak acid and therefore that neutral N: in methylamine (CH3NH2) must be a reasonably strong base. Thus, the relative electronegativities of N and O correctly predict that neutral N: will be a better base than neutral O: and that CH3OH2+ is a stronger acid than CH3NH3+. Note however that such predictions will hold only when the atoms being compared are in the same charge state, as is the case in the comparisons shown in Figures 5.4 and 5.5.
The second intrinsic property of elements that influences the acidity of their X–H bonds is their size. Elements within the same group (i.e., column) in the periodic table have the same number of valence shell electrons, but increase in size as we move down the table. As the atomic size increases, the X–H bond becomes longer, which weakens the bond and increases its acidity (Figure 5.6).

Figure 5.6 Example of effect of the size of the X atom on the acidity of X–H bonds in similar molecular contexts.
From the point of view of the conjugate base :X−, the negative charge becomes increasingly spread out as the size (i.e., volume) of the X atom increases. In general, as the negative charge in the conjugate base becomes more spread out, it becomes better stabilized and thus basicity decreases. So comparing down columns, increasing size of the X atom increases acidity of X–H and decreases basicity of :X−. As illustrated below, this trend also holds for acids of the BH+ type (Figure 5.7).

Figure 5.7 Example of the effects of atom size X on the acidity of +X–H bonds in similar molecular contexts (elements in the same column of the periodic table).
5.7 Atom Hybridization and Acid/Base Strength
Acidic X–H bonds are always covalent single bonds (σ bonds) arising from overlap of an atomic orbital on the X atom and the 1s orbital of H. The most common X atoms in organic molecules will be C, N, or O atoms involved in single, double or triple bonds. As discussed in Chapter 1, the σ-bonding orbitals associated with individual C, N, and O atoms are frequently envisioned as hybrid orbitals with varying degrees of s and p character, depending on the type of hybridization (sp, sp2, or sp3). Experimentally, it is observed that as hybridization of the orbital involved in an X–H bond changes from sp3 → sp2 → sp, the X–H bond becomes more acidic. This effect is shown for a series of similar molecules with acidic N–H and O–H bonds in the table (Table 5.4).
Table 5.4 Effect of X Atom Hybridization on Acidities of C–H, N–H, and O–H Acids (R = Methyl).

The explanation for this trend is that as the hybridization of an atom changes from sp3 → sp2 → sp, the percentage of “s” character increases from 25% to 33% to 50%. Recall that s orbitals are spherical in shape such that the electrons surround and overlap with the positively charged nucleus. In contrast, p orbitals have a bilobed “dumbbell” shape with a node at the nucleus such that the electrons are distributed further away from the nucleus. Thus, with increasing s character of the bonding orbital on the X atom (C, N, O), electrons in the X–H σ bond interact more strongly with the positively charged nucleus of X and less so with the bound proton. This results in a weakening of the X–H bond strength and an increased acidity (lower pKa).
5.8 Resonance Electronic Effects on Acid/Base Strength
In Chapter 1 we saw how resonance delocalization imparts partial double-bond character to amide bonds and affords special stabilization in aromatic ring systems like benzene. Not surprisingly, resonance delocalization of electrons plays a very important role in modulating the strengths of acids and bases as well. There are two major ways this can occur. The first is when lone pair electrons belonging to the basic X atom are delocalized and thus withdrawn into a larger π system (tending to make the X–H bond more acidic since :X− is stabilized). The second case is when another atom donates electrons into a π system that involves the basic X atom (tending to make the X–H bond less acidic since :X− is destabilized). We will see examples of both cases below, although the former scenario of electron withdrawal is more common than the latter.
As a first example of the effects of electron withdrawal through resonance delocalization, consider the trend in pKa values for several O–H containing functional groups (Figure 5.8). Each of the O–H bonds is potentially acidic, but the O atoms are attached in different molecular contexts allowing variable amounts of resonance electron delocalization and large differences in the relative acidities of the O–H bonds.

Figure 5.8 Example of effect of increasing resonance delocalization in functional groups with the same type of acidic X–H bond. In this figure and those that follow, atoms directly involved in resonance delocalization are shown in red.
Moving from left to right in this series the acids become stronger and the conjugate bases become weaker (less basic) as the negative charge becomes more spread out. In the conjugate base of methanol (i.e., the methoxide anion, CH3O−), the negative charge remains localized on the O atom and hence the basicity is similar to the basicity observed in HO− (pKa for H2O = 15.7).
Next in the series is phenol, the conjugate base of which is the phenoxide anion. The resonance hybrid structure of phenoxide (Figure 5.9) shows how the negative charge is distributed between the oxygen atom and the para- and ortho-carbon atoms by resonance delocalization. Since oxygen is more electronegative than carbon, the oxygen of phenoxide still carries the largest fraction of negative charge. However, the fractional charge on oxygen is much less in phenoxide as compared to methoxide and thus is less attractive to a proton (less basic). Comparing the pKa values (and recalling that each ΔpKa of 1 equals a 10-fold difference in relative basicity/acidity), the phenoxide anion is more than 100,000-fold less basic than the methoxide anion.

Figure 5.9 Resonance structures for phenoxide anion showing distribution of negative charge from O onto ortho- and para-carbon atoms of the phenyl ring.
Next in our series is acetic acid (pKa = 4.7), the conjugate base of which is the acetate anion (Figure 5.10). In acetate, the negative charge is delocalized onto two equally electronegative oxygen atoms, since the two resonance structures are identical. Each oxygen atom carries only 50% of the negative charge, which further stabilizes the anion and weakens the attraction to a proton. Comparing pKa values, this equal distribution of negative charge on two identical electronegative atoms makes the acetate anion more than 10 billion-fold less basic than methoxide anion and more than 100,000-fold less basic than phenoxide anion. This concept is extended further in the sulfonate anion (Figure 5.11) in which the negative charge is distributed equally among three oxygen atoms. Partial charge on each oxygen atom is reduced to just ~33%, affording the least “proton-seeking” conjugate base in the series.

Figure 5.10 Resonance structures of acetate anion showing equal distribution of negative charge on the two oxygen atoms.

Figure 5.11 Resonance structures for methylsulfonate anion with the negative charge distributed equally among three oxygen atoms.
To summarize, delocalization of electrons such that the negative charge is spread out among many atoms lowers the fractional charge on the X atom, which lowers its basicity and increases the acidity of its conjugate acid (X–H). Likewise, delocalizing electrons onto more electronegative atoms like oxygen is more effective at lowering basicity than delocalization onto less electronegative atoms like carbon.
Consider now some similar trends in acids based on nitrogen, that is, N–H acids (Figure 5.12). Consistent with the difference in electronegativity between N and O, each of the N–H acids in Figure 5.12 is substantially weaker than the corresponding O–H acids in Figure 5.8. As with the oxygen acids, we see the same trend of increasing acidity of the N–H bond (and decreasing basicity of :N−) with increased resonance delocalization. Note, however, that the magnitude of change in acidity (and basicity) is significantly greater for the N–H acids. For example, we observe a much larger difference in acidity for aniline and methylamine (~13 pKa units, or 1013-fold) than we saw between phenol and methanol (5 pKa units, or 105-fold). This difference between N and O bases can be understood in terms of electronegativity. Since N and C are closer in electronegativity than O and C (Table 5.3), the ortho- and para-carbon atoms in the aniline anion (PhNH−) share a larger proportion of the negative charge with N than the analogous carbon atoms in phenoxide share with O. Similarly, resonance delocalization in the acetamide anion results in >50% of the negative charge residing on the more electronegative O atom. This effect is greater than in the analogous case of acetate (Figure 5.10), where the charge is equally shared between the two oxygen atoms. Similar analyses can be extended to the other N–H acids in Figure 5.12 and for C–H type acids as well.

Figure 5.12 Comparison of acidities for neutral N–H acids shows same trend of increasing acidity with increasing electron delocalization, especially onto more electronegative atoms.
To summarize then, the stabilization of X− anions by resonance delocalization leads to a weaker base and to more acidic X–H bonds. Moreover, these effects are stronger in cases where the charge can be shared by atoms of greater electronegativity than the X atom.
5.8.1 Resonance Delocalization in BH+ Type Acids
Now we will consider the effects of resonance delocalization of BH+ type acids and their neutral conjugate bases (B:). Comparing the pKa values of two ammonium acids (R–NH3+) we see that the example where R = Ph is significantly more acidic than when R = Me (Figure 5.13). This suggests that the aromatic ring provides significant stabilization of the conjugate base, which in this case is the neutral molecule aniline. Inspection of a resonance hybrid of aniline shows how the lone pair electrons on nitrogen delocalize into the aromatic ring (Figure 5.14). With its electron density decreased, the N: atom in aniline is less basic than in methylamine and the corresponding anilinium ion (C6H5NH3+) is more acidic than the methylammonium ion (CH3NH3+).

Figure 5.13 Example of increased acidity of +N–H acids with increasing resonance electron delocalization in the conjugate base.

Figure 5.14 Resonance structures showing distribution of electron density among the N atom and the ortho- and para-carbon atoms of the phenyl ring.
In the examples above, we saw how delocalization of electrons from :X into a π system generally lowers the basicity of :X and makes the X–H bond more acidic. Now let’s examine some cases where resonance donation of electrons into a π system raises the basicity of :X and reduces acidity of the X–H bond of interest.
To illustrate this effect we will first compare the acidities of the guanidinium and iminium ions, which are BH+ type acids (Figure 5.15). In both cases, the N–H σ bond of interest involves an sp2 hybrid orbital on a nitrogen atom that is also involved in a C=N π bond. In the case of the iminium ion, the pKa simply reflects the intrinsic basicity of sp2-hybridized N. In the guanidinium ion, however, two N atoms are bound to carbon and each can contribute its lone pair electrons into the C=N π bond. The easiest way to see this is to examine a resonance hybrid structure of the guanidinium ion (Figure 5.16). As you can see, the effect of this resonance donation is to distribute the positive charge among all three nitrogen atoms. This greatly stabilizes the guanidinium ion, making it less prone to give up a proton and so much less acidic (pKa = ~13–14) than the iminium ion. From the perspective of the conjugate bases, we might say that guanidine is a much stronger base than an imine because it can more readily stabilize the positive charge that comes with attaining a proton.

Figure 5.15 Acid-base reactions and pKa values for the guanidinium ion (left) and an iminium ion (right).

Figure 5.16 Resonance structures showing distribution of positive charge among all three N atoms of the guanidinium ion.
Another example of the same effect is apparent in a comparison of the relative acidities of the aromatic imidazolium and pyridinium ions (Figure 5.17). Here again the acidic N–H bond involves an sp2 hybrid orbital that lies in the plane of the ring. However, the second nitrogen atom in the imidazole ring does contribute its lone pair electrons into the π system, to satisfy Hückel’s rule (Figure 5.18). This resonance donation makes imidazole more basic than pyridine. By the same token, pyridinium is a stronger acid because it cannot stabilize a positive charge in the way the imidazolium ion does—by sharing charge between its two nitrogen atoms. It might help to draw a resonance hybrid of the imidazolium ion to convince yourself of this!

Figure 5.17 Acid-base reactions and pKa values for the imidazolium ion (left) and the pyridinium ion (right).

Figure 5.18 Lone pairs of electrons in different types of orbitals have different basicities. Lone pairs in sp2-orbitals are basic and can be protonated. Lone pairs in p-orbitals that are part of an aromatic π system are not basic and do not become protonated.
5.9 Inductive Electronic Effects of Substituents on Acid/Base Strength
In the above sections we examined three factors that give rise to differences in relative acidities of X–H bonds within different functional groups. Most biological and drug molecules, however, have more than one functional group (substituents) and these may influence the electronic properties and acid/base properties of each other. The extent to which the properties are altered depends on the identity and placement of the substituents relative to one another, as well as on what types of bonds connect them. In this and the following two sections we examine the three main types of interactions between substituents: (1) inductive electronic effects, (2) combined resonance and inductive electronic effects, and (3) proximity effects.
As noted for resonance electronic effects, any structural feature that withdraws or pulls electron density away from a basic X atom decreases its basicity and increases the acidity of the corresponding X–H bond. Conversely, features that donate or push electron density toward a basic X atom increase its basicity and decrease the acidity of its corresponding X–H acid. While resonance effects result from direct delocalization of electron pairs into a conjugated π system, inductive effects result from induced polarization of σ bonds connecting the basic X atom of interest and a polarized substituent. The substituent is typically a functional group with a polarized bond, that is, a dipole moment. Inductive effects can be electron donating or electron withdrawing, depending on the direction of the dipole relative to the X–H bond (Figure 5.19).

Figure 5.19 Polarized bonds between atoms with different electronegativities may be inductively electron donating or electron withdrawing. In electron donating substituents the negative end of the dipole points toward the acidic X–H group, whereas in electron withdrawing substituents the positive end of the dipole points toward the acidic X–H group.
Recall that the directionality and magnitude of the dipole in a polarized bond is determined by the relative electronegativities of the bonded atoms. Of the atoms found most often in organic molecules, only H is less electronegative than C, while all of the common heteroatoms (X = N, O, S, F, Cl, Br, I) are more electronegative than C (Table 5.3). Thus, H–C bonds are polarized with slight greater electron density on carbon and thus are electron donating. In contrast, the greater electronegativity of the X heteroatoms makes X–C bonds polarized in the opposite direction and electron withdrawing.
As an example of the effects of the donating versus withdrawing substituents, consider the relative acidities of the O–H bond in acetic acid as compared to formic acid or chloroacetic acid (Figure 5.20). Three electron donating H–C bonds in the methyl group of acetic acid contribute electron density via σ bonds to the basic O, increasing its basicity and decreasing the acidity of the O–H bond in acetic acid (pKa ~4.8) relative to that in formic acid (pKa ~3.8). In contrast, replacement of methyl with chloromethyl reverses polarization of the substituent, which now pulls electron density away from the basic O, decreasing its basicity and increasing the acidity of the O–H bond in chloroacetic acid (pKa ~2.8) relative to that in acetic acid (pKa ~4.8).

Figure 5.20 Structures and pKa values for acetic acid as compared to either formic acid (left) or chloroacetic acid (right). Dipole moments are shown for specific substituents to illustrate inductive donating or withdrawing effects. In this figure and those that follow, the σ bonds connecting the basic O atom and the substituents being compared are shown as thick bonds.
Although alkyl groups are essentially the only electron donating substituents, there are a wide variety of electron withdrawing substituents of variable strength. To a first approximation, the polarization and consequent electron withdrawing effects will increase with increasing electronegativity of X in an X–C bond. Thus, the more electronegative atom fluorine is a stronger electron withdrawing substituent than chlorine, making fluoroacetic acid a stronger acid than chloroacetic acid (Figure 5.21).

Figure 5.21 Structures and pKa values for chloroacetic acid and fluoroacetic acid.
Not surprisingly, the presence of multiple polarized X–C bonds, whether single (e.g., F3C−, Cl3C−), or double, or triple bonds (e.g., O=C−, N≡C−, O=N−), increases the polarization and consequent inductive withdrawing effect of a substituent (Figure 5.22). Note that the nitrile (N≡C–) and nitro groups (NO2) in these examples exert their withdrawing effects via an inductive effect, since the basic O atom is not conjugated with the π bonds of these substituents.

Figure 5.22 Examples of increased inductive electron withdrawing effect of substituents with multiple X–C bonds on the acidity of the O–H bond.
Groups with double and triple bonds between C atoms (C=C or C≡C), although not polar themselves, are electron deficient and thus weakly pull electron density toward them through connecting σ bonds. This effect is evident in the case of the phenyl substituent in benzoic acid, which reduces the pKa slightly as compared to acetic acid. In contrast to the dramatic resonance effects observed in the cases of aniline and phenol, the effect in benzoic acid is more subtle and purely inductive since the aryl ring is connected to the basic O atom by two σ bonds rather than being conjugated with it (Figure 5.23).

Figure 5.23 Structures and pKa values for acetic acid and benzoic acid. The phenyl ring in the latter is weakly electron withdrawing by an inductive effect.
In the remainder of this section we will examine how the ionization state (charge state) of functional groups can dramatically affect their inductive withdrawing or donating effects. This phenomenon is highly relevant for proteins, and especially enzyme active sites where a number of ionizable groups are usually found in close proximity. Here we will observe these effects in the context of small molecules with multiple ionizable groups.
First, we consider the carboxylic acid functional group (COOH) and its ionized (anionic) form, the carboxylate anion (COO−). To evaluate the inductive effect of COOH, we can compare the pKa values of acetic acid with the dicarboxylic acids, succinic acid, and malonic acid (Figure 5.24). In the fully protonated diacids, the indistinguishable COOH groups are electron withdrawing, which we can see by noting that the lowest pKa value for either of the diacids is lower (more acidic) than acetic acid. However, once a proton is lost from a diacid the negatively charged carboxylate anion COO− exerts an electron donating effect on the remaining COOH group, decreasing its acidity substantially (higher second pKa compared to acetic acid). Another thing to note is that the inductive withdrawing effect of COOH in the fully protonated diacids exhibits much stronger distance dependence than does the donating effect of the carboxylate anion COO−. This is in line with what we have learned about distance dependence in Chapter 2 for ion-dipole or ion-ion interactions (less distance dependence) as compared to directional dipole-dipole interactions (greater distance dependence). The inductive withdrawing effect of the COOH group is akin to a dipole-dipole interaction and thus stronger in malonic acid than in succinic acid. By contrast, the effect of the carboxylate anion in the second deprotonation step can be thought of as an unfavorable charge-charge interaction.

Figure 5.24 Comparison of the acidity of acetic acid with two related dicarboxylic acids. The uncharged COOH groups act as inductive electron withdrawing groups (dipole arrows shown on malonic acid), but the negatively charged COO− group is inductively electron donating. The result of this is that the first pKa of the diacids is lower than for acetic acid while the second pKa is higher.
In contrast to the opposing effects of COOH and COO−, charged ammonium ions (−NR3+) and neutral amines (−NR2) are both electron withdrawing, but the charged forms are much more strongly withdrawing. These effects are apparent in comparing the acidities of two diammonium compounds with that of ethylammonium ion (Figure 5.25). The withdrawing effect of the ammonium group lowers the first pKa for the diacids relative to ethylamine, an effect that can be viewed as a combination of a strong inductive withdrawing effect and a repulsive ionic interaction present only in the doubly charged forms (and which is relieved by deprotonation). After the first proton is lost, the resulting neutral amino group retains a weakly withdrawing effect as evidenced by the greater acidity of the second ammonium group in 1,2,-diaminoethane (pKa = 9.9) as compared to ethylamine (pKa = 10.6). In 1,3-diaminopropane, however, the inductive withdrawing effect is not significantly transmitted over the four σ bonds separating the amino groups, so the second pKa value is only comparable to that of ethylamine.

Figure 5.25 Comparison of acidities of diammonium ions relative to the mono-ammonium ion of ethylamine.
The distance dependence of inductive electronic effects exemplified above with charged substituents can also be demonstrated by systematically moving a polarized bond further away from a basic atom X (Figure 5.26). In the mono-chlorobutanoic acids shown below, the inductive effect of the polarized C−Cl bond is strongest in chloroacetic acid (pKa = 2.8) and accordingly weaker as additional σ bonds are introduced between the withdrawing substituent and the basic O atom of the COOH function. Note that with just two σ bonds intervening, the withdrawing effect of the C−Cl bond is reduced significantly (pKa = 4.1), and with three σ bonds intervening, the effect is nearly eliminated (pKa = 4.5 vs. 4.8 for acetic acid).

Figure 5.26 The distance dependence of inductive effects is illustrated with a series of carboxylic acids bearing a polarized C–Cl bond at various distances from the carboxylic acid. Acetic acid (far right) serves as a reference compound lacking the withdrawing substituent.
5.10 Combined Inductive and Resonance Effects on Acid/Base Strength
When an acidic/basic functional group can interact with one or more substituents via a conjugated π system, a combination of inductive and resonance electron donating and withdrawing effects can occur. Resonance effects can arise from polarized substituents with π bonds such as C=O and S=O, which are resonance electron withdrawing, or from groups such as –NH2, –OR, and halides with lone pairs of electrons, which are resonance electron donating. Thus, the inductive and resonance effects of substituents like C=O and S=O act in concert to withdraw electron density from the conjugated X–H group. For substituents with lone pairs of electrons, however, inductive and resonance effects will oppose one another so that the overall effect will depend on which individual effect (inductive withdrawing or resonance donating) is stronger. When a substituent of either type is not fully conjugated with the acidic X–H group or not conjugated with it at all, the resonance contribution is substantially diminished and the overall effect will be dominated by the inductive effect of the substituent. We will now examine several examples to help illustrate these contrasting behaviors.
The effect of the nitro (NO2) group on the acidities of three different types of acidic functional groups is illustrated in Figure 5.27. The NO2 group is a π-containing substituent that is electron withdrawing via both inductive and resonance effects. Compared to the unsubstituted acids in the figure, the molecules that bear a nitro group have pKa values significantly lower. This indicates that the nitro-substituted analogs can better stabilize the deprotonated form of the acid (i.e., the conjugate base). Let us examine the source of this extra stabilization for the specific case of the para- and meta-substituted phenoxide anions.
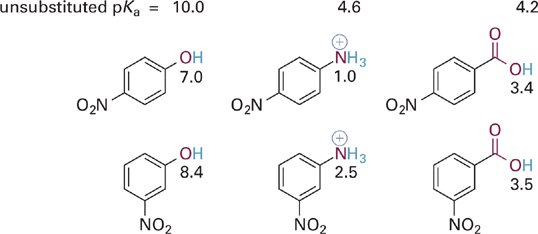
Figure 5.27 Illustration of the combined inductive and resonance effects of para- versus meta-nitro substitutions on the acidities of three different types of acidic groups—phenol, anilinium ion, and benzoic acids. The pKas of the unsubstituted parent compounds (without a nitro group) are given at the top for reference.
If we draw resonance hybrids of these two phenoxide anions, we observe that the para-substituted phenoxide possesses a particularly attractive resonance form (Figure 5.28). This is one in which a positive charge is positioned immediately adjacent to the anionic oxygen. We might draw an additional resonance form in which a C=O bond is formed between the atoms in question. What the resonance hybrid tells us is that electrons from the anionic oxygen atom can be fully delocalized throughout the ring and into the π system of the para-NO2 group as well, thus providing significant stabilization of the phenoxide anion. In contrast, the resonance hybrid for the meta-substituted analog does not allow for full delocalization of the negative charge on oxygen—one can “push electrons” from oxygen into the ring or from the ring onto the nitro group, but not both at the same time. The important thing to take away from this analysis is that the electron withdrawing effect of a meta-NO2 group (and of meta-substituted π-type substituents in general) involves mainly inductive and not resonance effects. In contrast, substitution at the para (or ortho) position with such groups involves a stronger electron withdrawing effect resulting from a combination of inductive and resonance effects.

Figure 5.28 Comparison of key resonance structures for the para- and meta-nitro substituted phenoxide anions.
Next, let us consider the case of para- or meta-substitution with a methoxy group (OCH3) in an analogous series of acids (Figure 5.29). The methoxy group with its lone pair electrons is considered to exert a fairly strong resonance electron donating effect but also a fairly strong inductive electron withdrawing effect on account of the electronegativity of oxygen. In the examples at hand, the effect of para-methoxy substitution is electron donating (higher pKa values compared to the unsubstituted systems), indicating that resonance electron donation overwhelms the inductive withdrawing effect for para-substitution. In contrast, the overall effects of methoxy in the meta position is electron withdrawing (lower pKa values compared to unsubstituted systems), indicating that resonance electron donation is either not effective at this position or is much smaller than the inductive electron withdrawing effect.

Figure 5.29 Comparison of the differences in the combined inductive and resonance effects of para- versus meta-methoxy substitutions on the acidities of three different types of acidic groups. The pKas of the unsubstituted parent compounds are given at the top for reference.
The above noted effect of the para-methoxy group on pKa can be understood by examining key resonance structures for the corresponding conjugate bases (Figure 5.30). Resonance donation of a lone pair of electrons into the aromatic π system places a negative charge in proximity to other negatively charged or electron-rich atoms. The net contribution of these resonance forms then is to destabilize the conjugate bases slightly, leading to the observed increases in pKa for the corresponding acid forms. The effect of meta-methoxy substitution is primarily inductive withdrawal of electrons, and is distance dependent—thus larger for compounds with fewer σ bonds between the substituent and the acidic X–H bond (Figure 5.31).

Figure 5.30 Comparison of key resonance structures for conjugate bases of three different para-OCH3 acids. The ΔpKa values refer to change in pKa compared to unsubstituted parent compound (ΔpKa = pKa-subst – pKa-parent)

Figure 5.31 Meta-substituents primarily increase acidity through inductive electron withdrawal through σ bonds (bold bonds) and exhibit typical distance dependence.
The effects of other substituents bearing lone-pairs on electronegative atoms are demonstrated below in the context of phenolic and benzoic acids (Figure 5.32). The halogen atoms chlorine and fluorine are found to be slightly (for F) or moderately (for Cl) electron withdrawing at the para or ortho position, but most strongly withdrawing at the meta position. Why might this be so? The explanation is that these atoms exert a relatively modest resonance electron donating effect, less than the methoxy group but still significant. This resonance effect of the halogens is strongest at the ortho and para positions, for the same reason as it is in methoxy. Thus, the resonance effect partially offsets the dominant inductive withdrawing effect of the halogens when located at the ortho or para positions. At the meta position, resonance effects are minimal (as is generally the case) and so the strong inductive effect dominates.

Figure 5.32 Comparison of the combined inductive and resonance effects of para- and meta-halides and amino groups on the acidities of acidic groups. The pKa values of the unsubstituted parent compounds are given at the top for reference and the pKa values of the substituted analogs are given near each structure.
Finally, comparison of the acidities for the meta- and para-aminobenzoic acids in Figure 5.32 shows that the intrinsically more basic N atom is overall electron donating in both positions, but as expected, is much more strongly donating at the para position. This is due to the full conjugation of the amino group with the π system of the COO− group, combined with a very weak inductive withdrawing effect. Amino groups are among the few substituents in which resonance electron donation dominates over inductive withdrawing effects, even at the meta position. Methyl groups are one of the few inductive electron donating groups, and since they have no resonance effect, a methyl (or other alkyl group) is electron donating at both the para and meta positions. This effect is similar to the amino group, albeit with a much smaller magnitude of electron donation.
To summarize, resonance electron withdrawal or donation is always strongest in the para and ortho positions because only in these positions can the substituent be fully conjugated with the acidic functional group. The four general categories of combined effects described in this section are as follows:
1. Substituents such as nitro with π bonds conjugated to aromatic rings, which are electron withdrawing by a combination of resonance and inductive effects. This effect is strongest at the para position.
2. Substituents such as the halides, which are strong inductive electron withdrawing groups at all positions, but are most withdrawing at the meta position. This is because of a weak and opposing electron donating resonance effect in the para or ortho position.
3. Other electronegative substituents such as OMe or OH are inductive electron withdrawing groups but have lone pairs of electrons that participate in resonance electron donation. These countering effects lead to an overall donating effect at the para and ortho positions, but an overall withdrawing effect at the meta position.
4. Less electronegative substituents like amino groups (:NR2) have accordingly weaker inductive withdrawing properties and so act primarily as resonance electron donors. The effect is strongest at the para and ortho positions.
5.11 Proximity and Through-Space Effects on Acid/Base Strength
In addition to the through-bond electronic effects of substituents on acid/base strength discussed in Sections 5.8 through 5.10, substituents in close proximity to acidic groups can also alter the acid/base behavior via through-space hydrophobic effects or direct hydrogen bonding interactions.
This first example compares the effects of increasing the number of hydrophobic substituents in close proximity to neutral X–H versus charged BH+ type acids (Figure 5.33).

Figure 5.33 Comparison of the opposite effects of increasing numbers of hydrophobic substituents in close proximity to an uncharged X–H acid (top) versus a charged BH+ acid (bottom).
In the first series, successive replacement of H atoms by −CH3 groups leads to slight decreases in acidity of the uncharged alcohol O–H, as would be predicted for addition of more inductively electron donating methyl groups. In the second series, however, only the first replacement of H for ethyl (−C2H5) gives rise to a significant decrease in acidity for the positively charged +NH acid of the alkylammonium ion. The second replacement gives a further slight decrease in acidity, but the third replacement reverses the trend, indicating that another force is acting in opposition to the inductive donating effect of the alkyl groups.
If we imagine these molecules in aqueous solution we can better understand what is going on (Figure 5.34). As each hydrogen substituent is replaced by a hydrophobic ethyl group, stabilizing hydrogen bonds between water molecules and the charged BH+ acid are lost. Thus, the change in the local environment of the +N–H acid from highly polar to more hydrophobic has the effect of destabilizing the charged acid form relative to the neutral N–H base. As a result of the opposing inductive and proximity effects, we find that primary, secondary and tertiary alkylammonium ions often have very similar pKa values in aqueous solution.

Figure 5.34 Increasing numbers of hydrophobic substituents decreases the number of stabilizing hydrogen bonds to water in the ionic BH+ acid, which decreases the stability of the acid making it lose its proton more easily (increases its acidity).
Similar effects are now recognized to operate with neutral O–H acids as well. As the microenvironment becomes more hydrophobic, the anionic conjugate base is disfavored relative to the neutral acid form. Thus, in the case of O–H acids, the local environment effect acts in concert with the inductive electron donation of the alkyl substituents, leading to significantly decreased acidity with each additional alkyl substituent.
Another common proximity effect occurs in molecules capable of forming intramolecular hydrogen bonds to an acidic functional group (Figure 5.35). In acetylsalicylic acid, the O-linked ester group provides a traditional inductive electron withdrawing effect that increases the acidity of the carboxylic acid function, lowering its pKa to 3.5 (as compared to pKa 4.2 for benzoic acid). In salicylamide, however, two factors are responsible for the significant reduction in pKa to ~8.1 (from ~ 10 for phenol). First, the carbonyl (C=O) of the amide provides a modest resonance electron withdrawing effect. Second, the formation of a hydrogen bond (dashed line) between the amide N–H donor and the basic O: atom provides for stabilization of the conjugate base (phenoxide anion O−).

Figure 5.35 Arrow showing inductive electron withdrawing effect of the ester in acetylsalicylic acid (left) and combination of direct hydrogen bond and resonance electron withdrawing effects of the amide group in salicylamide (right).
Now consider the case of salicylic acid, where both an acidic phenol and carboxylic acid are present in close proximity (Figure 5.36). The overall effect is that the carboxylic acid O–H is a stronger acid than benzoic acid (pKa = 3.0 vs. 4.2), while the phenolic O–H is a dramatically weaker acid as compared to phenol (pKa 13.4 vs. 10). How can we understand these effects? Although the resonance donating effect of an ortho O–H would be expected to decrease the acidity of the COOH (contrary to what is observed), the O–H group is perfectly positioned to act as a hydrogen bond donor to the basic O of the carboxylic acid (Figure 5.36). This proximity effect is much more important than the resonance effect, and reduces the basicity of the carboxylate anion (thus increasing COOH acidity). However, once the carboxylate anion has formed, the phenolic proton will be favorably hydrogen bonded to it. Moreover, to undergo a second deprotonation will involve the energetically unfavorable formation of two proximal negative charges. Overall, the combination of these forces raises the pKa for the second deprotonation to pKa = 13.4, making the phenolic O–H group in salicylic acid >1000-fold less acidic than the O–H group in phenol (pKa = 10).

Figure 5.36 Successive loss of protons in salicyclic acid. Hydrogen bonding between phenolic O–H and O of COOH increases acidity of COOH, but decreases acidity of phenolic O–H in the monobasic anion (middle structure).
5.12 The Henderson–Hasselbalch Relationship and Acid/Base Equilibria as a Function of pH
As noted at the outset of this chapter, the charge states of acidic and basic groups can have important influences on the reactivities of drug molecules, on their aqueous versus lipid solubility, and on the specificity of the binding interactions with their target molecules. Thus, it is important to review the principles of acid/base equilibria that dictate how the charge state of the individual acidic and basic functional groups in a molecule, as well as of the whole molecule, change as a function of pH.
Starting with the definition of the equilibrium acid dissociation constants from Section 5.5:

Recall that when an acid or base is dissolved in a solution that is well buffered at a specific pH (i.e., [H3O+] is constant), the relative concentrations of base [A−] and acid [HA] (i.e., [A−]/[HA]) is determined by the ratio of the equilibrium dissociation constant Ka to the constant [H3O+].
Taking the negative log of the above equation for Ka gives the Henderson–Hasselbalch equation:

Substituting pKa = – log Ka and pH = – log [H3O+] gives the simpler relationship:

Rearranging shows the amount of A− relative to HA is simply determined by the difference between the pH and the pKa:
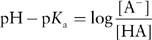
By definition, the pKa for an acid is equal to the pH where [HA] = [A−], that is, the acid is 50% dissociated. At pH values below the pKa, the acid is present at higher concentrations than its conjugate base ([HA] > [A−]) and at pH values above the pKa, the acid is present at lower concentrations than its conjugate base ([HA] < [A−]). Figure 5.37 depicts this schematically.

Figure 5.37 Relationship between fraction or percent of the acid (or base) forms of a functional group and the difference between the pH and pKa.
From this relationship it is clear that for neutral H–A acidic groups, the uncharged HA state predominates at pH values below its pKa while the charged anionic base A− predominates at pH values above the pKa.
The same equation can be derived from the equilibrium dissociation constant for BH+ acids:

where once again the acid form predominates at pH values below its pKa while the base form predominates at pH values above the pKa. Keep in mind that whereas H–A acids are charged (anionic) in the conjugate base form (A−), BH+ acids are charged (cationic) in the acid form and neutral in the conjugate base form (B:). The power of the Henderson–Hasselbalch equation is that it allows one to readily predict the charge state of drugs and specific functional groups at various pH values. The environment a drug might encounter ranges from quite acidic (e.g., the stomach, pH ~1–3) to mildly acidic (pH ~5 in the lysosome) to neutral (pH ~7.4 in the cytosol of a cell). Being able to predict the protonation/charge state of a drug in these various environments allows one to understand and make predictions about drug solubility, permeability, and interactions with their target molecules.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


