CHAPTER 7 Abnormalities of the red cell membrane
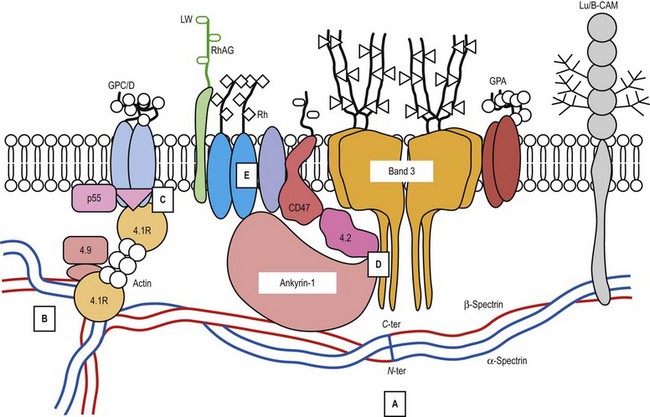
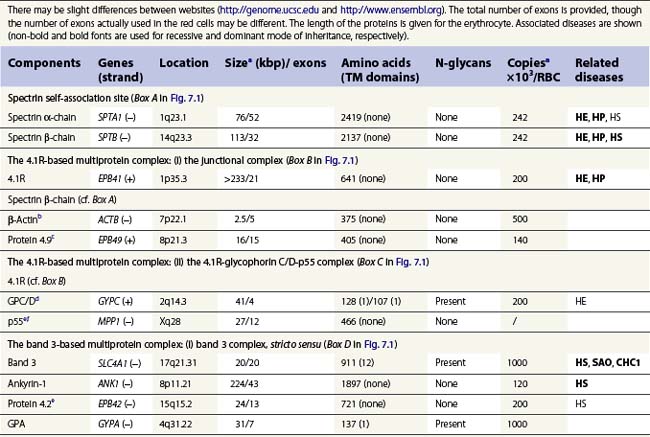
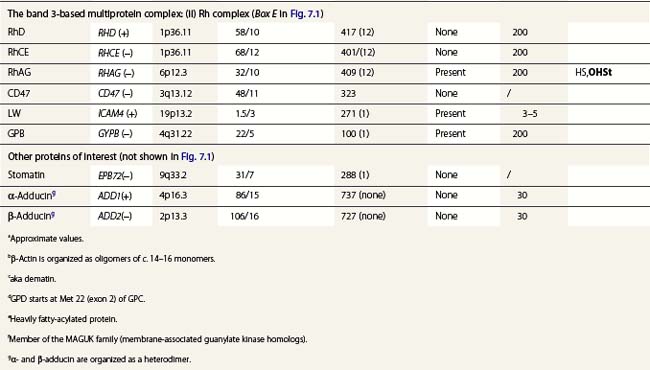
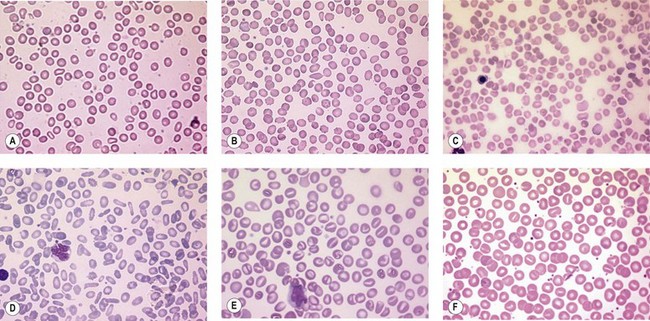
A number of hereditary hemolytic anemias result from mutations affecting the quality and/or amount of proteins that belong to the red cell membrane, its skeleton, or the attachment systems (nexuses) of the latter to the former. Most proteins participate in complexes (Fig. 7.1). They play a role in erythrocyte resilience and elastic deformability, either mechanically, through the skeleton and its attaching systems, or osmotically, through a variety of transporters and pumps. Major proteins and their genes are listed in Table 7.1. The ever increasing number of mutations, too numerous to detail here, have given insight into the function of such protein domains and the regulatory regions of some genes. A selection of abnormally-shaped red cells is shown in Fig. 7.2.
Hereditary spherocytosis
Hereditary spherocytosis (HS) is the most common genetic disorder of the red cell membrane in Western countries. Its incidence has been estimated as 1 in 2000 live births and there is a wide spectrum of clinical severity. In typical cases the hemolytic anemia is moderate, with an increased reticulocyte count a reticulocytosis, intermittent jaundice, gallstones and splenomegaly. Severe cases are rare and may cause death in utero or shortly after birth. In contrast, patients with mild HS may be over 60 years of age when diagnosed. Parvovirus B19 infection commonly occurs. Blood films show a variable percentage of spherocytes. The diagnosis relies on an increased percentage of hyperdense cells and a reduction in osmotic resistance, and on a number of tests, including polyacrylamide gel electrophoresis of the red cell membrane proteins in the presence of sodium dodecylsulfate (SDS-PAGE). The main treatment is splenectomy, though the need for this should be carefully weighed owing to its complications, namely severe infections and a statistically significant increase in thromboembolic accidents.1 Transfusions may be necessary.
ANK1 gene mutations
Ankyrin-1 is encoded by ANK1.2 It connects the skeleton to band 3, i.e. the anion exchanger-1. Approximately 60% of HS are due to ANK1 gene mutations and have reduced ankyrin-1. HS due to ANK1 mutations is relatively severe and has a dominant inheritance pattern, although de novo mutations may occur. Homozygosity is bound to be lethal. (One case has been recently described, however.) This may not be evident due to the elevated reticulocyte count, as young cells have a higher ankyrin-1 content. Spectrin α- and β-chains, and protein 4.2, interacting with ankyrin-1, are secondarily decreased.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


