CHAPTER 41 Wassif S. Wassif; James E. East CHAPTER OUTLINE Multiple endocrine neoplasia type 1 Multiple endocrine neoplasia type 2 Other familial syndromes associated with multiple endocrine neoplasia METABOLIC CHANGES IN MALIGNANCY ENDOCRINE SEQUELAE OF TUMOURS AND THEIR TREATMENT Reproductive consequences of therapy Tumours may exert metabolic effects on the host via a wide range of mechanisms. Some of the metabolic derangements related to tumours are directly related to a hormone or other substance that the tumour secretes. Such hormone secretion can be appropriate to the cell line from which the tumour originates or may be inappropriate or ‘ectopic’. In the last decade, molecular genetic techniques have helped characterize tumours and explain how neoplastic tissues can secrete hormones that are not typically associated with them. Metabolic derangements can also arise non-specifically, in the absence of hormone secretion or immunological phenomena, for example owing to tumour burden compromising host metabolism or the effects on the host of rapid cell turnover. Autoimmune reactions evoked by tumour antigens may also give rise to systemic effects and are increasingly being implicated in the causation of various tumour-related or paraneoplastic, syndromes. The recognition and characterization of the antibodies to shared tumour and host antigens that play a prominent role in paraneoplastic syndromes have advanced their use in both diagnosis and treatment. Scientific advances have rapidly opened up new possibilities for the management of tumours, even in their advanced stages, where formerly only observation and palliation was possible. Clinical biochemistry has a key role in both diagnosis and monitoring treatment. Neuroendocrine cells can be identified by their production of a neurotransmitter, neuromodulator or neuropeptide hormone. Neuroendocrine tumours (NETs) are distinguished by their ability to secrete peptides causing characteristic endocrine syndromes. The gastroenteropancreatic system has diffuse neuroendocrine components and NETs most commonly arise from these tissues, more than 50% being carcinoid tumours. Carcinoids are NETs that arise from enterochromaffin cells, principally in the intestine (~ 80% in the ileum), the main bronchi and, rarely, other tissues including the ovaries, thymus, pancreas and thyroid. Carcinoid tumours all show a similar pattern of neuroendocrine expression and may contain or secrete amines, peptides or prostaglandins. The carcinoid syndrome is the clinical result of systemic release of these substances, particularly serotonin. The incidence of carcinoid tumours found at post-mortem has been estimated to be as high as 1 in 150, whereas carcinoid syndrome only occurs in approximately 1 in every 50 000 individuals, indicating that most tumours appear to be are non-secretory. The products of primary intestinal carcinoids are metabolized in the liver prior to release into the systemic circulation: the development of carcinoid syndrome in patients with a gastrointestinal tumour therefore indicates the presence of hepatic metastases. Carcinoid tumours at bronchial (~10% of the total) and other sites secrete products directly into the systemic circulation and can be associated with carcinoid syndrome without the presence of metastases. The main clinical features of carcinoid syndrome are flushing and diarrhoea (Box 41.1). The diarrhoea tends to be secretory in type and weight loss is common. Patients who flush repeatedly may, in time, develop a cyanotic telangiectatic appearance, which persists. Wheezing may occur, caused by transient bronchoconstriction, which is most commonly associated with bronchial carcinoids. Hypertension is not typically a feature of carcinoid syndrome: hypotension is more common. Carcinoid syndrome is also associated with fibrosis; the bowel, retroperitoneum (and hence ureters), lungs and heart can all be affected. Fibrosis is thought not to be linked directly to serotonin but to mitogenic growth factors that drive fibroblast proliferation. Tryptophan is a precursor of both serotonin and nicotinic acid. Thus, in carcinoid syndrome, excess production of serotonin may lead to nicotinamide deficiency, manifesting as pellagra with dermatitis of sun-exposed areas. Carcinoid crisis has been described in certain circumstances including induction of anaesthesia, liver biopsy and following combination chemotherapy. It is manifest as extreme flushing, explosive diarrhoea, labile blood pressure and cardiac rhythm irregularities, including asystole. The rate limiting step in the biosynthesis of serotonin is the hydroxylation, by tryptophan hydroxylase, of tryptophan to 5-hydroxytryptophan (5HTP); this is subsequently decarboxylated to yield 5-hydroxytryptamine (serotonin, 5HT) (see Fig. 41.1). 5-Hydroxytryptamine is stored in neurosecretory granules or secreted into the bloodstream. After secretion, some is taken up into platelets. Oxidative deamination, catalysed by monoamine oxidase and aldehyde dehydrogenase, inactivates 5HT to 5-hydroxyindoleacetic acid (5HIAA), which is excreted in the urine. FIGURE 41.1 Pathway of synthesis and metabolism of 5-hydroxytryptamine. The normal excretion of 5-HIAA is < 50 μmol/24 h. In patients with typical carcinoids, 99% of metabolized 5HT and 5HTP are excreted as 5HIAA. Urinary 5HIAA excretion is typically in the range 150–1500 μmol/24 h, generally exceeds 500 μmol/24 h and occasionally is as high as 3000 μmol/24 h. Carcinoid tumours of the colon and rectum do not contain the hydroxylase or decarboxylase enzymes and so do not form 5HTP or 5HT. A small number of patients have ‘atypical’ tumours. These patients excrete large amounts of 5HTP and 5HT in their urine. It is believed that these tumours, usually bronchial, are deficient in dopa decarboxylase and cannot convert 5HTP to 5HT so that the former is secreted into the bloodstream. Some of the 5HTP is converted to 5HT and subsequently to 5HIAA in extrarenal sites; some is decarboxylated in the kidneys and excreted into the urine as 5HT, and some escapes decarboxylation and is excreted directly into the urine. Thus, patients with atypical carcinoid tumours have a marked increase in 5HTP and 5HT excretion and only a moderate increase in 5HIAA excretion. Even in those patients whose tumours produce predominantly 5HTP, urinary 5HIAA constitutes 50–60% of total urinary 5-hydroxyindoles and increased values are found in almost all patients. The most sensitive indicator of the turnover of serotonin and its metabolites is measurement of 5HIAA in a 24 h urine sample. A number of dietary substances can interfere in the measurement of 5HIAA and should be avoided for three days prior to and during the collection of the urine sample (see Box 41.2). The test is not completely specific and a negative result is to be expected in patients with non-secretory tumours and those of the hindgut that do not produce 5HT. Chromogranin A (CgA) is a 48 kDa protein distributed in dense core granules of neuroendocrine cells, the highest plasma concentrations being found in patients with carcinoid tumours. It has been reported to have a sensitivity of 75–85% and a specificity of 84–95% in the diagnosis of carcinoid syndrome. It is not dependent upon serotonin secretion and is therefore particularly useful in patients with non-secretory or atypical carcinoids and as its concentration reflects tumour burden, it can be used as a marker for assessing response to treatment. False positive results can be seen in hepatic and renal failure, atrophic gastritis, proton pump inhibitor therapy, and inflammatory bowel disease. Prostatic cancers, which may contain a significant neuroendocrine component, myeloma, exercise, trauma and hypertension may also increase CgA concentrations. In isolated carcinoid tumours, where surgical excision is a management option, anatomical localization may be useful. Conventional imaging modalities include computed tomography (CT), magnetic resonance imaging (MRI) and positron emission spectroscopy (PET). A more specific option is the use of somatostatin receptor scintography, e.g. using 111In-DTPA-pentetreotide (Octreoscan™), which binds to somatostatin receptors on the tumour. Circumscribed tumours may be removable by surgical resection. For patients with significant hepatic metastatic disease, radiofrequency ablation or hepatic embolization, alone or in combination with intra-arterial chemotherapy (chemoembolization), while not curative, may provide significant palliation. Somatostatin analogues have been used effectively in the management of the symptoms of carcinoid syndrome. Multiple endocrine neoplasia (MEN) syndromes are disorders characterized by pathological hyperfunction of two or more endocrine organs. They have been classified into two distinct disorders: MEN type 1 (MEN1) and MEN type 2 (MEN2) with MEN2 further subdivided into three main variants (see Box 41.3). Two distinct genetic defects contribute to tumourigenesis in MEN syndromes: in MEN1, inactivation (loss of function) of a tumour suppressor gene is thought to be responsible, while MEN2 is caused by overactivation (gain of function or overexpression) of a proto-oncogene. A tumour suppressor gene restrains cell proliferation and tumours are stimulated by inactivating mutations or deletions in such a gene, which render the gene product either absent or nonfunctional. In contrast, a proto-oncogene (the normal unmutated version of an oncogene) can be converted to an oncogene, which can cause cell proliferations or deregulated cell growth when overexpressed. Multiple endocrine neoplasia type 1 (MEN1) typically becomes manifest after the first decade of life, with most men and women developing symptoms in the fourth and third decades, respectively. Typically, MEN1 tumours appear two decades earlier than isolated endocrine tumours. Primary hyperparathyroidism (HPT) is the most frequent endocrinopathy in MEN1 and is the most common reason for the disease to come to the attention of physicians. It occurs in 90% of affected individuals between 20 and 25 years of age, rising to nearly 100% by the age of 50 years. However, MEN1 itself is rare and accounts for only 2–4% of patients with HPT. There is no sex difference in MEN1 prevalence compared with a ratio of 3:1 females:males in sporadic hyperparathyroidism. Hyperparathyroidism is often found during the second decade of life during screening of immediate family members of an index patient with proven MEN1. The pathological features are those of diffuse or asymmetrical hyperplasia, with all four glands being involved (although perhaps not all to the same degree) or with multiple adenomas. The investigation and management of HPT is discussed in Chapter 6. Gastroenteropancreatic (GEP) neuroendocrine tumours are the second most common tumours in MEN1, with some 60% of patients being affected. They usually present between the second and fifth decades, unless diagnosed earlier by screening. Approximately half of the tumours are gastrinomas. The Zollinger–Ellison syndrome (ZES) is severe, intractable, multiple and recurrent peptic ulcer disease caused by a gastrin secreting tumour, which can be located in the duodenum or pancreas. The majority of gastrinomas associated with MEN1 are in the duodenum, where they are often small (< 0.5 cm in diameter) and multiple. Since gastrin secreting cells are not normally found in the duodenum or pancreas, gastrinomas in these sites should be regarded as ectopic and potentially malignant, irrespective of their histological grade. The clinical syndrome does not differ from that seen with non-MEN1 gastrinomas. The extreme hypersecretion of gastric acid may be associated with inactivation of pancreatic lipase, resulting in fat malabsorption and steatorrhoea. Hyperparathyroidism in MEN1 can exacerbate hypergastrinaemia. The diagnosis can be made by demonstrating increased gastric acid secretion with simultaneously elevated plasma gastrin concentrations. Secretin infusion (2 U/kg) may cause augmented gastrin production in those patients whose basal values are equivocal. Excellent treatments are now available for the medical management of peptic ulcer disease using histamine H2 receptor antagonists or proton pump inhibitors. Other GEP NETs include insulinomas, vasoactive intestinal peptide secreting tumours (VIPomas) and glucagonomas. Their clinical features are discussed further in Chapter 12. Chromogranin A is useful, particularly as a marker of midgut tumours, in the investigation of GEP NETs. Cocaine- and amfetamine-regulated transcript (CART), is a 116 amino acid peptide widely distributed in nervous and endocrine tissues and its physiological roles may include regulation of feeding and response to psychological stress. It is produced by various islet cell tumours and is of particular use as a marker in the investigation of NETs of pancreatic origin. Measurement of a screening panel of gut hormones may also be useful. The true prevalence of anterior pituitary tumours in MEN1 is unclear. Between 30% and 50% of patients with MEN1 develop anterior pituitary tumours. Whilst some are non-functional, approximately 60% secrete prolactin, 25% secrete growth hormone and 5% secrete ACTH. Occasionally, excess secretion of GH and cortisol in MEN1 is the result of ectopic secretion of GHRH and ACTH, respectively, e.g. by a pancreatic islet or carcinoid tumour. It is important to identify such patients in order to ensure appropriate therapy. The investigation and management of pituitary tumours in patients with MEN1 is similar to that for other pituitary tumours and is discussed in detail in Chapter 18. Carcinoid tumours occur more frequently with MEN1 than in the general population; some 10% of MEN1 patients are affected. In contrast to sporadic carcinoid tumours, which are predominantly derived from the midgut and hindgut, MEN1 carcinoid tumours are usually derived primarily from the foregut. Foregut carcinoids rarely secrete serotonin, peptide hormones or calcitonin and are usually considered as clinically non-functional. Adrenal cortical lesions are common in MEN1: 20–40% of patients are affected, mostly with bilateral tumours. However, the majority are non-functional, clinically silent and rarely require treatment. Cushing and Conn syndromes are rare in MEN1. MEN1 is the result of an inactivating mutation in the MEN-1 gene, a tumour suppressor gene located on chromosome 11q13. It consists of 10 exons and encodes a 610-amino acid nuclear protein termed menin. Menin interacts with diverse groups of transcription factors and coregulators, including JunD, suggesting a role in gene transcription, DNA replication and cell cycle control. Genetic mapping studies indicate somatic loss of heterozygosity in accord with the ‘two hit’ hypothesis. The first hit is a genetic mutation rendering the subject heterozygous for the MEN-1
Metabolic effects of tumours
INTRODUCTION
NEUROENDOCRINE TUMOURS
Carcinoid tumours
Clinical presentation
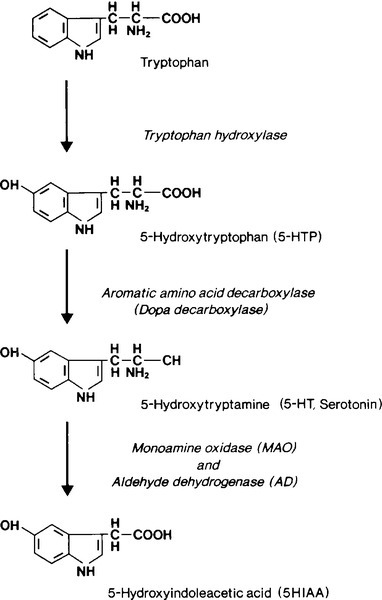
Metabolism of serotonin
Laboratory investigation
Diagnostic imaging
Treatment
MULTIPLE ENDOCRINE NEOPLASIA
Multiple endocrine neoplasia type 1
Parathyroid disease
Gastroenteropancreatic neuroendocrine tumours
Pituitary tumours
Foregut carcinoid tumours
Adrenal tumours
Tumourigenesis in MEN1
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


