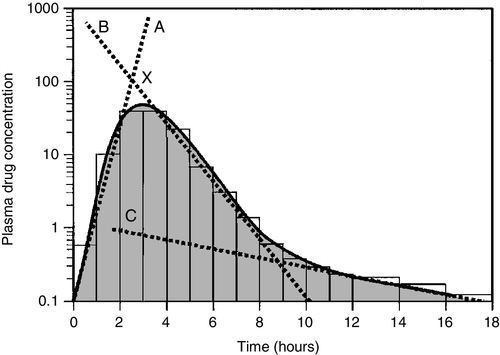
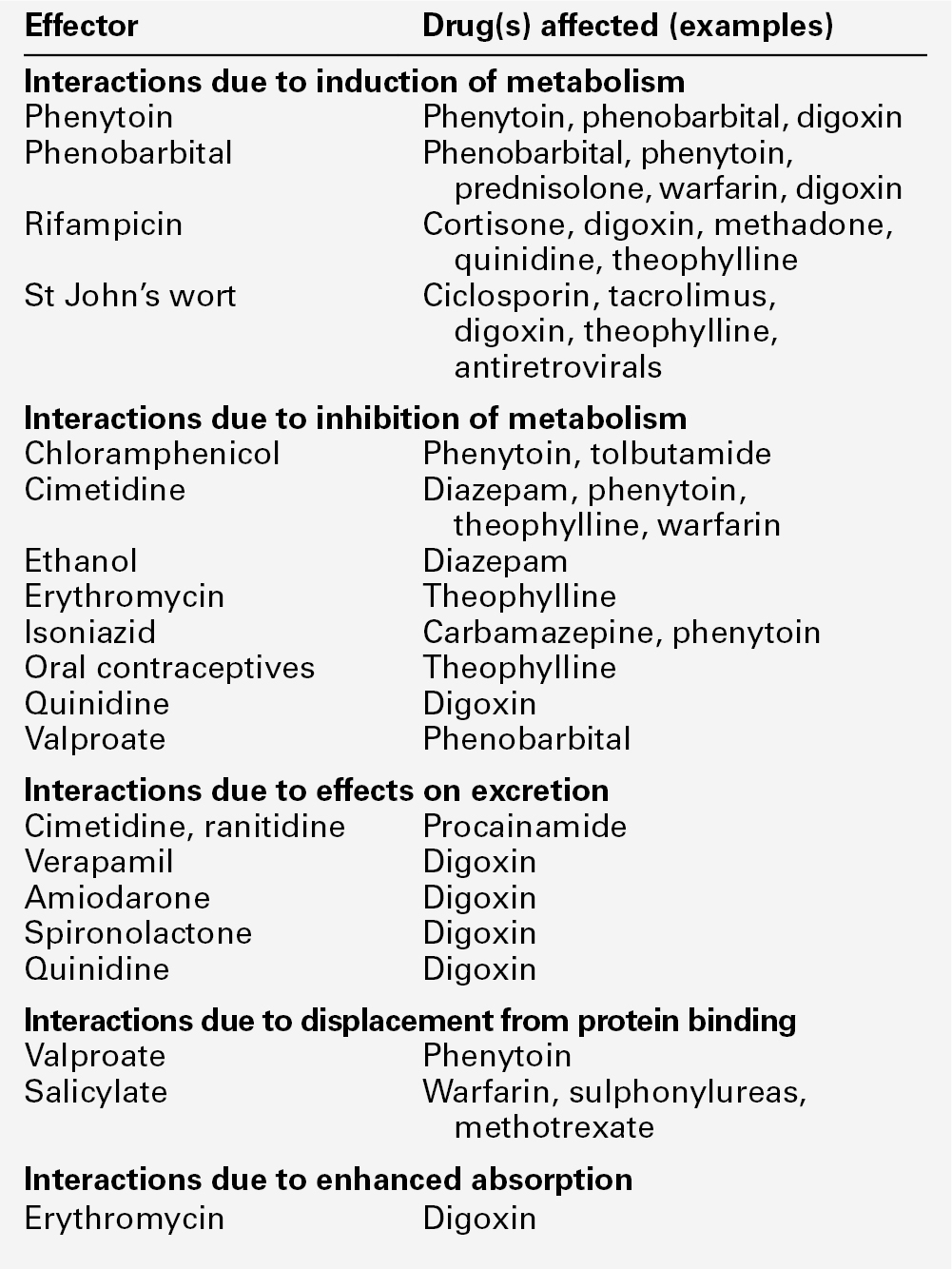
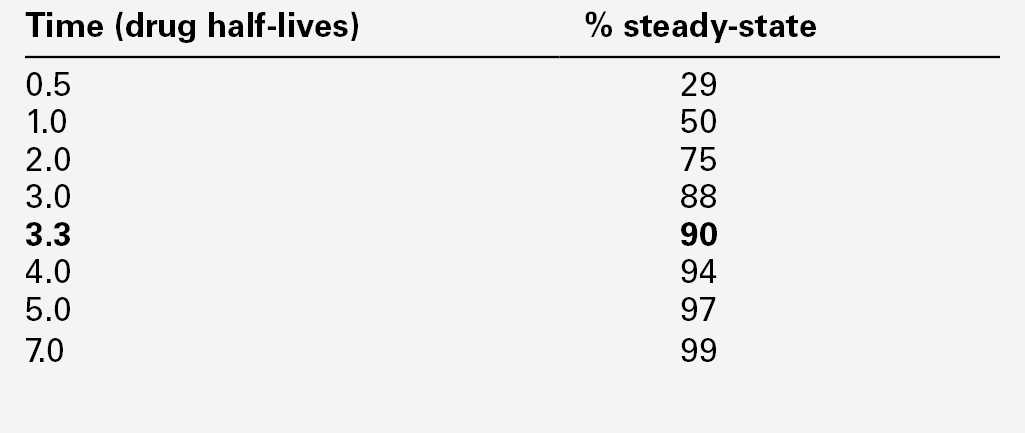
CHAPTER 39 CHAPTER OUTLINE Pharmacokinetics and pharmacodynamics Which drugs should be measured? USE OF THERAPEUTIC DRUG MONITORING Relevant clinical interpretation PROVISION OF A THERAPEUTIC DRUG MONITORING SERVICE PHARMACODYNAMIC MONITORING, BIOMARKERS AND PHARMACOGENETICS Analgesic/anti-inflammatory drugs Antiarrhythmics and cardiac glycosides Anticonvulsants (antiepileptics) Antidepressants and antipsychotic drugs The aim of therapeutic drug monitoring (TDM) is to aid the clinician in the choice of drug dosage in order to provide the optimum treatment for the patient and, in particular, to avoid iatrogenic toxicity. It can be based on pharmacogenetic, demographic and clinical information alone (a priori TDM), but is normally supplemented with measurement of drug or metabolite concentrations in blood or markers of clinical effect (a posteriori TDM). Measurements of drug or metabolite concentrations are only useful where there is a known relationship between the plasma concentration and the clinical effect, no immediate simple clinical or other indication of effectiveness or toxicity and a defined concentration limit above which toxicity is likely. Therapeutic drug monitoring has an established place in enabling optimization of therapy in such cases. Before discussing which drugs to analyse or how to carry out the analyses, it is necessary to review the basic elements of pharmacokinetics and pharmacodynamics. Essentially, pharmacokinetics may be defined as what the body does to drugs (the processes of absorption, distribution, metabolism and excretion), and pharmacodynamics as what drugs do to the body (the interaction of pharmacologically active substances with target sites (receptors) and the biochemical and physiological consequences of these interactions). The processes involved in drug handling are summarized in Figure 39.1, which also indicates the relationship between pharmacokinetics and pharmacodynamics, and will now be discussed briefly. For a more mathematical treatment, the reader is referred to the pharmacokinetic texts listed in Further reading. FIGURE 39.1 Processes involved in drug handling. The first requirement for a drug to exert a clinical effect is obviously for the patient to take it in accordance with the prescribed regimen. Patients are highly motivated to comply with medication in the acute stages of a painful or debilitating illness, but as they recover and the purpose of medication becomes prophylactic, it is easy for them to underestimate the importance of regular dosing. ‘Compliance’ is the traditional term used to describe whether the patient takes medication as prescribed, though ‘adherence’ has become the preferred term in recent years as being more consistent with the concept of a partnership between the patient and the clinician. ‘Compliance’, however, remains in widespread use. In chronic disease states such as asthma, epilepsy or bipolar disorder, variable adherence is widespread. The fact that a patient is not taking a drug at all is readily detectable by measurement of drug concentrations, although variable adherence may be more difficult to identify by monitoring plasma concentrations. Once a drug has been taken orally, it needs to be absorbed into the systemic circulation. This process is described by the pharmacokinetic parameter bioavailability, defined as the fraction of the absorbed dose that reaches the systemic circulation. Bioavailability varies between individuals, between drugs and between different dosage forms of the same drug. In the case of intravenous administration, all of the drug goes directly into the systemic circulation and bioavailability is 100% by definition, but different oral formulations of the same drug may have different bioavailability depending, for example on the particular salt or packing material that has been used. Changing the formulation used may require dosage adjustment guided by TDM to ensure that an individual’s exposure to drug remains constant. Other routes of absorption such as intramuscular, subcutaneous or sublingual may exhibit incomplete bioavailability. The total amount of drug absorbed can be determined from the area under the plasma concentration/time curve (see Fig. 39.2). FIGURE 39.2 Concentration/time curve for a drug administered by any route other than intravenously. Line A represents the absorption phase; line B indicates the distribution half-life and line C the elimination half-life. At point X, absorption, distribution and metabolism are all in progress. The areas of the rectangles can be summed to estimate the total amount of drug absorbed, the ‘area under the curve’ (AUC) (hatched area). When a drug reaches the bloodstream, the process of distribution to other compartments of the body begins. The extent of distribution of a drug is governed by its relative solubility in fat and water, and by the binding capabilities of the drug to plasma proteins and tissues. Drugs that are strongly bound to plasma proteins and exhibit low lipid solubility and tissue binding will be retained in the plasma and show minimal distribution into tissue fluids. Conversely, high lipid solubility combined with low binding to plasma proteins will result in wide distribution throughout the body. The relevant pharmacokinetic parameter is the volume of distribution, which is defined as the theoretical volume of a compartment necessary to account for the total amount of drug in the body if it were present throughout the compartment at the same concentration found in the plasma. High volumes of distribution thus represent extensive tissue binding. After reaching the systemic circulation, many drugs exhibit a distribution phase during which the drug concentration in each compartment reaches equilibrium, represented by Line B in Figure 39.2. This may be rapid (~ 15 min for gentamicin) or prolonged (at least six hours for digoxin). There is generally little point in measuring the drug concentration during the distribution phase, especially if the site of action of the drug for clinical or toxic effects lies outside the plasma compartment and the plasma concentration in the distribution phase is not representative of the concentration at the receptor. Between-patient variations in volume of distribution caused by differences in physical size, the amount of adipose tissue and the presence of disease (e.g. ascites) can significantly weaken the relationship between the dose of drug taken and the plasma concentration in individual patients and hence increase the need for concentration monitoring. When a drug has been completely distributed throughout its volume of distribution, the pharmacokinetics enter the elimination phase (line C in Fig. 39.2). In this phase, the drug concentration falls owing to metabolism of the drug (usually in the liver) and/or excretion of the drug (usually via the kidneys into the urine or via the liver into the bile). These processes are described by the pharmacokinetic parameter clearance, which is a measure of the ability of the organs of elimination to remove active drug. The clearance of a drug is defined as the theoretical volume of blood that can be completely cleared of drug in unit time (cf. creatinine clearance, see Chapter 7). Factors affecting clearance and hence the rate of elimination include body weight, body surface area, renal function, hepatic function, cardiac output, plasma protein binding and the presence of other drugs which affect the enzymes of drug metabolism including alcohol and nicotine (tobacco use). Pharmacogenetic factors may also have a profound influence on clearance. Rate of elimination is often expressed in terms of the elimination rate constant or the elimination half-life, which is the time taken for the amount of drug in the body to fall to half its original value and is generally easier to apply in clinical situations. Both the elimination rate constant and the elimination half life can be calculated from the clearance and the volume of distribution (see Pharmacokinetic texts in Further reading). With some drugs (e.g. phenytoin), the capacity of the clearance mechanism is limited, and once this is saturated there may be large increments in plasma concentrations with relatively small increases in dose. This phenomenon makes these drugs very difficult to use safely without access to concentration monitoring. Many endogenous constituents of plasma are carried on binding proteins in plasma (e.g. bilirubin, cortisol and other hormones), and drugs are also frequently bound to plasma proteins (usually albumin and α1-acid glycoprotein). Acidic drugs (e.g. phenytoin) are, in the main, bound to albumin, whereas basic drugs bind to α1-acid glycoprotein and other globulins. The proportion of drug bound to protein can vary from zero (e.g. lithium salts) to almost 100% (e.g. mycophenolic acid). The clinical effect of a drug and the processes of metabolism and excretion are related to the free concentration of a drug, with drug bound to protein effectively acting as an inert reservoir. Variations in the amount of binding protein present in plasma can thus change the total measured concentration of the drug in the blood without necessarily changing the free (active) concentration, and this is a further reason why measured plasma concentrations may not relate closely to clinical effect. Other drugs or endogenous substances that compete for the same binding sites on protein will also affect the relationship between free and bound drug. For these reasons, it has been suggested that the free concentration of a drug rather than the total (free + protein-bound) concentration should be measured by a TDM service, at least for those drugs exhibiting significant protein binding, e.g. phenytoin. Despite the apparent logic of this idea, it has not been widely adopted because of methodological difficulties and continuing controversy about whether the theoretical benefits are realized in clinical practice. It is nonetheless important to be aware of the effects of changes in protein binding when interpreting total drug concentrations in plasma, especially: • when there is a highly abnormal binding protein concentration in plasma; for example, in severe hypoalbuminaemia • when a pathological state (e.g. uraemia) results in the displacement of drug from the binding sites. The discussion above has outlined the pharmacokinetic factors that govern the relationship between dose and the drug concentration in the plasma or at the active site, and has indicated how these may vary between patients to produce poor correlation between the dose prescribed and the effective drug concentration at the site of action. In general, once steady-state has been reached, plasma concentrations in the individual should exhibit a constant relationship to the concentration at the site of action governed by the distribution factors discussed above, but this may not be the case if blood supply to target tissues is impaired (e.g. for poorly vascularized tissues or a tumour that has outgrown its blood supply (cytotoxic drugs) or a site of infection that is not well perfused (antibiotics)). Pharmacodynamics is the study of the relationship between the concentration of drug at the site of action and the biochemical and physiological effect. The response of the receptor may be affected by the presence of drugs competing for the same receptor, the functional state of the receptor or pathophysiological factors such as hypokalaemia. Interindividual variability in pharmacodynamics may be genetic or reflect the development of tolerance to the drug with continued exposure. High pharmacodynamic variability severely limits the usefulness of monitoring drug concentrations as they are likely to give a poor indication of the effectiveness of therapy. The above discussion provides a basis for determining which drugs are good candidates for therapeutic drug monitoring. As stated at the beginning of this chapter, the aim of TDM is the provision of useful information that may be used to modify treatment. For this reason, it is generally inappropriate to measure drug concentrations where there is a good clinical indicator of drug effect. Examples of this are the measurement of blood pressure during antihypertensive therapy; glucose in patients treated with hypoglycaemic agents; clotting studies in patients treated with heparin or warfarin, and cholesterol in patients treated with cholesterol-lowering drugs. While plasma concentration data for such drugs are valuable during their development to define pharmacokinetic parameters and dosing regimens, TDM is not generally helpful in the routine monitoring of patients. It may have a limited role in detecting poor adherence or poor drug absorption in some cases. However, where there are no such clinical markers of effect or where symptoms of toxicity may be confused with those of the disease being treated, concentration monitoring may have a vital role. Therapeutic drug monitoring is useful only for drugs that have a poor correlation between dose and clinical effect (high pharmacokinetic variability). Clearly, if dose alone is a good predictor of pharmacological effect, then measuring the plasma concentration has little to contribute. However, clinically useful TDM does require that there is a good relationship between plasma concentration and clinical effect. If drug concentration measurements are to be useful in modifying treatment, then they must relate closely to the effect of the drug or its toxicity (or both). This allows definition of an effective therapeutic ‘window’ – the concentration range between the minimal effective concentration and the concentration at which toxic effects begin to emerge – and allows titration of the dose to achieve concentrations within that window. Demonstration of a clear concentration–effect relationship requires low between-individual pharmacodynamic variability (see above), the absence of active metabolites that contribute to the biological effect but are not measured in the assay system and (usually) a reversible mode of action at the receptor site. Reversible interaction with the receptor is required for the intensity and duration of the response to the drug to be temporally correlated with the drug concentration at the receptor. Many drugs have active metabolites, and some drugs are actually given as pro-drugs, in which the parent compound has zero or minimal activity and pharmacological activity resides in a product of metabolic transformation. For example, mycophenolate mofetil is metabolized to the active immunosuppressant mycophenolate. It will be clear that useful information cannot be obtained from drug concentration measurements if a substantial proportion of the drug’s effect is provided by a metabolite that is not measured and whose concentration relationship to the parent compound is undefined. In some cases (e.g. amitriptyline/nortriptyline), both drug and metabolite concentrations can be measured and the concentrations added to give a combined indication of effect, but this assumes that drug and metabolite are equally active. In other cases (e.g. carbamazepine and carbamazepine 10,11-epoxide), the active metabolite is analysed and reported separately. Therapeutic drug monitoring is most valuable for drugs which have a narrow therapeutic window. The therapeutic index (therapeutic ratio, toxic-therapeutic ratio) for a drug indicates the margin between the therapeutic dose and the toxic dose – the larger, the better. For most patients (except those who are hypersensitive), penicillin has a very high therapeutic ratio and it is safe to use in much higher doses than those required to treat the patient, with no requirement to check the concentration attained. However, for other drugs (e.g. immunosuppressives, anticoagulants, aminoglycoside antibiotics and cardiac glycosides), the margin between desirable and toxic doses is very small, and some form of monitoring is essential to achieve maximal efficacy with minimal toxicity. The criteria for TDM to be clinically useful are summarized in Box 39.1. The list of drugs for which TDM is of proven value is relatively small (Box 39.2). Phenytoin and lithium are perhaps the best and earliest examples of drugs that meet all the above criteria and for which TDM is essential. The aminoglycoside antibiotics, chiefly gentamicin and tobramycin, also qualify on all counts. A number of other drugs that are frequently monitored fail to meet one or more of the criteria completely, and the effectiveness of TDM as an aid to management is therefore reduced. The evidence for the utility of monitoring many drugs is based more on practical experience than well-designed studies. However, for newer agents such as the immunosuppressants and antiretroviral drugs, there is good evidence supporting the benefits of TDM in improving clinical outcomes. (Further information is given in Individual Drugs, below.) Once the narrow range of drugs for which TDM can provide useful information has been defined, it should not be assumed that TDM is necessary for every patient on these drugs at every visit. For TDM to be used for maximum patient benefit and optimal cost-effectiveness, six important criteria must be satisfied each time a sample is taken. These are summarized in Box 39.3 and will now be discussed briefly. The first essential for making effective use of any laboratory test is to be clear at the start what question is being asked. This is particularly true for TDM requests, and much time, money and effort is wasted on requests where the indication for analysis has not been clearly defined. If the question is uncertain, the answer is likely to be unhelpful. The two main reasons for monitoring drugs in blood are to ensure effective therapy and to avoid toxicity. Effective therapy requires that sufficient drug reaches the drug receptor to produce the desired response (which may be delayed in onset). Where a drug is prescribed and the desired effect is not achieved, this may or may not be due to insufficient dosage, since there may be other reasons (individual idiosyncrasy, drug interactions etc.) for the lack of effect. Those at the extremes of age – neonates and the very elderly – have metabolic processes that render them differently susceptible even to weight-adjusted doses. For example, in neonates, the metabolism of theophylline is qualitatively as well as quantitatively different from that in older children. In the very elderly, there may be considerable alterations in absorption and also renal clearance. Drug interactions may also produce a reduced clinical effect for a given dose. In patients on long-term therapy, once a steady-state concentration that produces a satisfactory clinical effect has been obtained, this concentration can be documented as an effective baseline for the individual patient. If circumstances subsequently change, then changes in response can be related back to both the dose and the plasma concentration of the drug. This is particularly important for psychotropic drugs. ‘Baseline’ concentrations may however change over time as disease processes develop, or gradually with increasing age or changes in drug metabolism. The avoidance of iatrogenic toxicity is probably the most pressing case for the practice of TDM. The aim is to ensure that drug (or metabolite) concentrations are not so high as to produce symptoms/signs of toxicity. Since a narrow therapeutic index is a prerequisite for drugs suitable for TDM, it is inevitable that toxicity will occur in a small proportion of patients, even with all due care being exercised. Toxicity can never be diagnosed solely from the plasma drug concentration, and it must always be considered in conjunction with the clinical circumstances since some patients will show toxicity when their concentrations are within the generally accepted therapeutic range and others will tolerate concentrations outside the range with few or no ill-effects. The advantage of regular monitoring is that such circumstances may be recorded and the range for that individual adjusted accordingly. There are two main factors that may lead to inappropriately high plasma drug concentrations. The first is an inappropriate dosing regimen, either due to a single gross error or (more often) a gradual build-up of plasma concentration either because the dose is slightly too high for the individual or because of the development of hepatic or renal insufficiency. The second factor leading to toxicity is pharmacokinetic drug interactions. Patients are often treated with more than one drug, which can interfere with each other’s actions in a number of ways, for example: • displacement from protein binding sites • competition for hepatic metabolism • induction of hepatic metabolizing systems • competition for renal excretory mechanisms. Examples of the more commonly encountered drug interactions involving drugs measured for TDM purposes are given in Table 39.1. There are, however, numerous other examples and the analyst faced with an unusual response or an inappropriate plasma concentration should seek a full drug history to assist in determining the explanation. This history should include specific enquiry about the use of alternative therapies, since many herbal and other non-pharmaceutical remedies (which may not be immediately mentioned by patients) may have significant effects on drug concentrations due to the induction of drug metabolizing enzymes. St John’s wort (Hypericum perforatum), a perennial herb with bright yellow flowers, is one example. It has been shown to induce the hepatic drug-metabolizing enzymes CYP3A4 and CYP2B6, and hence reduce the steady-state concentrations of many drugs. Therapeutic drug monitoring is particularly useful in confirming toxicity when both under- and overdosage with the drug in question give rise to similar clinical features, for example arrhythmias with digoxin, or fits with phenytoin, and where high drug concentrations give rise to delayed toxicity, for instance high aminoglycoside concentrations, which may give rise to irreversible ototoxicity if prompt action is not taken. Where a patient is known to be suffering from toxicity and the plasma drug concentration is high, monitoring is often required to follow the fall in concentration following cessation of treatment. For example, in anticonvulsant overdose, it is important for the clinician to know when drug concentration(s) are likely to reach the therapeutic range, since reinstatement of therapy will then be required to prevent seizures. Other important reasons for therapeutic drug monitoring include guiding dosage adjustment in clinical situations in which the pharmacokinetics are changing rapidly (e.g. neonates, children or patients in whom hepatic or renal function is changing) and defining pharmacokinetic parameters and concentration–effect relationships for new drugs. The use of TDM to assess adherence (compliance) is to some extent controversial. Clearly, assessment of adherence could provide justification for monitoring every drug in the pharmacopoeia, at enormous cost and for little clinical benefit. Further, application of TDM in this situation is not simple. A patient with a very low or undetectable drug concentration is usually assumed to be non-compliant, but the situation is much less clear when a concentration that is only slightly low (based on population data) is found. Is the patient non-compliant or are other factors (such as poor absorption, induced metabolism or altered protein binding) leading to the low concentration? Much harm may be done in these situations by assumptions of poor adherence to therapy. Adherence can be assessed in other ways, by tablet-counting, supervised medication in hospital or the use of carefully posed questions that are non-judgemental, for example ‘How often do you forget to take your tablets?’ Such approaches are likely to be more effective than TDM in detecting and avoiding poor adherence to therapy, though TDM may have a role in patients with poor symptom control, who deny poor adherence despite careful questioning and interventional reinforcement. Proper interpretation of TDM data depends on having some basic information about the patients and their recent drug history. The importance of this requirement has led many laboratories to design specific request forms for TDM. These are now being superseded by computerized request ordering systems that allow collection of essential items of information at the time of making the request, although it remains important to keep the requesting interface user-friendly and not too complex. Intelligent system design is necessary. Basic information requirements for TDM requests are summarized in Table 39.2. TABLE 39.2 Information requirements for TDM requests The vast majority of TDM applications require a blood sample. In general, serum or plasma can be used, although for a few drugs that are concentrated within red cells, for example, ciclosporin, whole blood is preferable as concentrations are higher and partition effects are avoided. The literature is poor on the differentiation between serum and plasma samples and careful selection of sample tubes is necessary to avoid interferences by anticoagulants, plasticizers, separation gels etc, either in the assay system or by absorption of sample drug onto the gel or the tube, reducing the amount available for analysis. If in doubt, serum collected into a plain glass tube with no additives is usually safest. Haemolysis should be avoided, as it may cause in vivo interference if the drug is concentrated in erythrocytes, or in vitro interference in immunoassay systems. For drugs that are minimally protein bound, there need be no restrictions on the use of tourniquets; however, stasis should ideally be avoided for drugs which are highly bound to albumin, e.g. phenytoin. Care should also be taken to avoid contamination with local anaesthetics, for example lidocaine, which are sometimes used before venepuncture. Where intravenous therapy is being given, care must be taken to avoid sampling from the limb into which the drug is being infused. Urine is of no value for quantitative TDM. Saliva may provide a useful alternative to avoid venepuncture (especially in children) or when an estimate of the concentration of free (non-protein-bound) drug is required. Saliva is effectively an in vivo ultrafiltrate and concentrations reflect plasma free drug concentrations quite well for drugs that are essentially unionized at physiological pH. Salivary monitoring is unsuitable for drugs that are actively secreted into saliva (e.g. lithium) and drugs that are strongly ionized at physiological pH (e.g. valproic acid, quinidine) as the relationship between salivary and plasma concentrations becomes unpredictable. Careful collection of the sample is required, and the mouth should be thoroughly rinsed with water prior to sampling. Sapid (taste related) or masticatory (chewing on an elastic band) stimulation of saliva flow is used to increase volume. The mucoproteins in saliva make the sample difficult to handle, and centrifugation is usually necessary to remove cellular debris. These and other problems have meant that salivary analysis has not been widely adopted for routine TDM. It is normally necessary for the patient to be at steady-state on the current dose of drug, i.e. when absorption and elimination are in balance and the plasma concentration is stable. This is true except when suspected toxicity is being investigated, when it is clearly inappropriate to delay sampling until steady-state has been reached. The time taken to attain the steady-state concentration is determined by the plasma half-life of the drug, and the relationship between the number of half-lives which have elapsed since the start of treatment and the progress towards steady-state concentrations is shown in Table 39.3. It is frequently stated that five half-lives must elapse before plateau concentrations are achieved, unless loading doses are employed when they are achieved much more rapidly. As shown in Table 39.3
Therapeutic drug monitoring
INTRODUCTION
Pharmacokinetics and pharmacodynamics
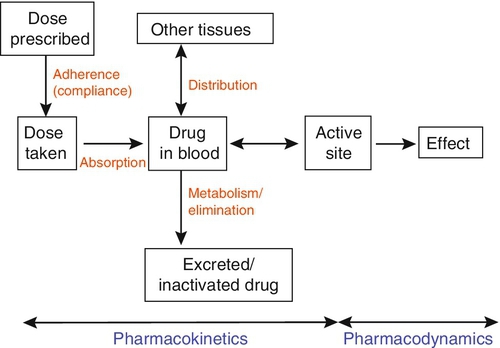
Adherence
Absorption
Distribution
Elimination (metabolism and excretion)
Protein binding
Pharmacodynamic factors
Which drugs should be measured?
USE OF THERAPEUTIC DRUG MONITORING
Appropriate clinical question
Accurate patient information
Essential
Desirable
Patient
Name
Weight
Age
Renal/hepatic function
Gender
Hospital/health system ID number
Pathology/clinical details
Problem
Reason for request
(e.g. poor response, ?toxic)
Therapy
Drug of interest dose formulation and route of administration duration of therapy date/time last given
Other drugs – list all
Appropriate sample
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


