OUTLINE
Responsibility for Providing Quality Drug Products
I. DEFINITIONS
A. Stability: In USP Chapter 〈1191〉 Stability Considerations in Dispensing Practice, stability is defined as “the extent to which a product retains, within specified limits, and throughout its period of storage and use (i.e., its shelf-life), the same properties and characteristics that it possessed at the time of its manufacture” (1). Chapter 〈1191〉 recognizes five types of stability: chemical, physical, microbiologic, therapeutic, and toxicologic (1).
Stability is determined by appropriate stability testing. The purpose of stability testing is to provide evidence on how the quality of a drug substance or drug product varies with time under the influence of a variety of environmental factors such as temperature, humidity, and light and to establish a shelf life for the drug product and recommended storage conditions.
B. Physical properties: These are the properties of drugs and dosage forms that we can see or test by physical means. Is the drug a solid, a liquid, or a gas? Is it dissolved, suspended, or emulsified, or is it adsorbed to the surface of a container? When a physical change occurs, the same drug or chemical is still present, but its physical state is altered. Pharmaceutical examples of physical changes include a drug precipitating out of solution; a drug adsorbing to the walls of a polyvinyl chloride (PVC) container; and two solid drugs forming a liquid eutectic mixture when triturated together in a mortar.
1. From a pharmaceutical viewpoint, there are both desirable and undesirable physical changes. When we make a drug solution, we want the physical change of the solid drug dissolving in the chosen solvent. In contrast, when we have an intravenous drug solution, it is unacceptable, and perhaps lethal, for the drug to precipitate out of solution.
2. Chapter 〈1191〉 of the USP gives the following criteria for acceptable levels of physical stability:“The original physical properties, including appearance, palatability, uniformity, dissolution, and suspendability, are retained” (1).
C. Chemical properties: The chemical properties of drugs are those manifested by the drug’s particular molecular structure. When a chemical change occurs, the original drug molecule is no longer present.
1. Recall from your study of general chemistry some of the types of reactions that occur with inorganic molecules. For example,
a. Acid-base neutralization reactions, such as:
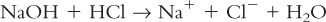
b. Oxidation-reduction reactions, such as:
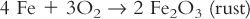
c. Displacement reactions, such as:
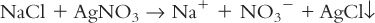
d. Release of gas, such as:
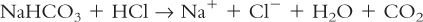
2. Most drugs are complex organic molecules, and chemical changes that occur with them are often more complicated than the simple types shown here; your study of organic and medicinal chemistry has provided you with the detailed knowledge you need to understand and to anticipate many of these changes.
3. Usually we buy drugs of a desired structure, and any change in that structure is undesirable. We then say that degradation or decomposition has occurred. Occasionally, however, we make use of a chemical change to prepare a desired drug preparation, as in making finely divided sulfur and potassium sulfide from solutions of zinc sulfate and sulfurated potash when compounding Zinc Sulfide Topical Suspension, also known as White Lotion:
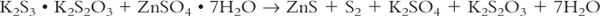
4. Chapter 〈1191〉 of the USP gives the following criteria for acceptable levels of chemical stability:“Each active ingredient retains its chemical integrity and labeled potency, within the specified limits” (1).
D. Microbiologic properties: Drug products should be free of microbiologic contamination and should resist any microbial growth.
1. Though certain products, such as parenterals and ophthalmics, are required to be sterile, all drug products should be free of microbiologic contamination.
2. For drug products that are labile to the growth of microorganisms introduced during use, preservatives should be added. Preservatives are discussed in Chapter 16, Antimicrobial Preservatives.
3. Chapter 〈1191〉 of the USP gives the following criteria for acceptable levels of microbiologic stability: “Sterility or resistance to microbial growth is retained according to the specified requirements. Antimicrobial agents that are present retain effectiveness within the specified limits” (1).
E. References: Table 37.1 gives a list of references that contain helpful information on compatibility and stability of drugs and drug products and preparations.
II. RESPONSIBILITY FOR PROVIDING QUALITY DRUG PRODUCTS
A. Providing the public with quality drug products is the joint responsibility of regulatory agencies such as the Food and Drug Administraiton (FDA), pharmaceutical manufacturers, and pharmacists.
B. Standards and guidelines for manufacturing, marketing, handling, and dispensing these products are set by the following:
1. Federal and state governments through agencies such as the FDA and state boards of pharmacy
2. Nonprofit groups and commissions, such as the United States Pharmacopeial Convention (USP), the National Association of Boards of Pharmacy, and the Joint Commission on Accreditation of Healthcare Organizations

3. Professional associations, such as the American Pharmacists Association and the American Society of Health-System Pharmacists
C. Responsibility of the pharmacist
1. In its General Information Chapter 〈1191〉 Stability Considerations in Dispensing Practice, the USP clearly states that it is the pharmacist’s responsibility to ensure that drug products provided to patients meet acceptable criteria of stability (1). Chapter 〈1191〉 is recommended reading for all pharmacy students, pharmacists, and pharmacy technicians.
a. Chapter 〈1191〉 outlines those factors that affect product stability. The pharmacist should be conscious of these factors when handling and storing drug products (1).
(1) Ingredients, whether therapeutically active or pharmaceutically necessary, can affect the stability of drug substances and dosage forms.
(2) Environmental factors that can reduce stability include exposure to adverse temperatures, light, humidity, oxygen, and carbon dioxide.
(3) Dosage form factors that influence drug stability include particle size (emulsions and suspensions), pH, solvent system composition (water and overall polarity), compatibility of anions and cations, solution ionic strength, primary container, chemical additives, and molecular binding between drugs and excipients.
b. To help ensure that quality, stable pharmaceutical products are dispensed and used, the USP recommends that pharmacists do the following (1):
(1) Watch for and comply with expiration dates, rotate stock, and use older products first.
(2) Store drugs and drug products under recommended environmental conditions.
(3) Observe products for evidence of instability.
(4) Properly handle drugs and drug products that require extemporaneous manipulation.
(5) Package products using recommended containers and closures.
(6) Educate patients about the proper storage, use, and disposal of drug products and preparations.
2. The pharmacist shares a responsibility with pharmaceutical manufacturers for the stability of manufactured drug products, and pharmacists are encouraged to report to the manufacturer and to the FDA any problems with packaging, labeling, or evidence of instability in manufactured drug products. The report to the FDA can be done easily by going to its Web site at www.fda.gov. The home page has a selection for product problem reporting. A report may be submitted online or by telephone, or the paper report form can be downloaded and faxed to FDA or sent using a postage-paid addressed form. As of October 2008, the FDA program for reporting drug product problems is called MedWatch, but the agency changes or reorganizes its programs from time to time, so it is best to check their Web site for current information.
3. While the USP recognizes five types of stability, this chapter concentrates on chemical and physical stability because when these are maintained, the others follow. Microbiological stability has been discussed in previous chapters (see Chapter 17, Antimicrobial Preservations, and the Chapters in Part 6, Sterile Dosage Forms and their Preparation).
4. In the following discussion, the stability and compatibility of both manufactured products and extemporaneously compounded drug preparations are considered, but the major emphasis is on drug preparations made or manipulated by the pharmacist and pharmacy technician.
D. FDA regulation of product stability
1. The International Conference on Harmonization (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use is a unique project that brings together the regulatory authorities of Europe, Japan, and the United States and experts from the pharmaceutical industry in the three regions to discuss scientific and technical aspects of product registration. The purpose is to make recommendations on ways to achieve greater harmonization in the interpretation and application of technical guidelines and requirements for product registration in order to reduce or obviate the need to duplicate the testing carried out during the research and development of new medicines. Within this context, ICH has developed a number of guidance documents for the stability testing of pharmaceutical products. The activities of the ICH are divided into four major topic categories, one being all the things related to chemical and pharmaceutical quality assurance of drug products. Within this category, ICH has published six guidance documents related to the stability of pharmaceutical products. These guidances include the following:
a. Stability Testing of New Drug Substances and Products (2)
b. Photostability Testing of New Drug Substances and Products (3)
c. Stability Testing for New Dosage Forms (4)
2. The “Stability Testing for New Dosage Forms” is especially important for the pharmacist interested in stability testing (4). This guidance gives recommendations on what should be done regarding stability testing of new dosage forms of known drugs that are already registered. A new dosage form is considered to be a drug product that is a different pharmaceutical product type that contains the same active substance as included in the existing approved drug product. These include products of different administration route (e.g., oral to rectal), new specific functionality/delivery systems (e.g., immediate release tablet to modified release tablet), and different dosage forms of the same administration route (e.g., capsule to tablet, solution to suspension).
III. PHYSICAL CHANGES
A. The USP (1) defines physical stability as the assurance that the product retains its original physical properties, including appearance, palatability, uniformity, dissolution, and suspendability, during its shelf-life.
B. Liquefaction of solid ingredients
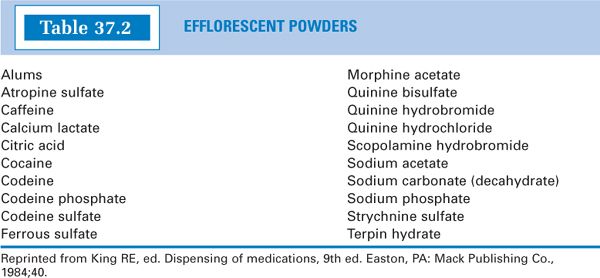
1. Efflorescent powders: These powders contain water of hydration that may be released when the powders are triturated or when stored in an environment of low relative humidity. The water liberated when the drug or chemical is triturated may cause the powders to become damp or pasty. If water is released to the atmosphere because of low relative humidity, the drug loses its crystallinity and becomes powdery. Furthermore, if water of hydration is given off, a given weight of the resulting powder no longer contains the same amount of drug.
a. For examples of efflorescent drugs, see Table 37.2.
b. Strategies for handling these drugs include the following:
(1) Store and dispense these powders in tight containers.
(2) The anhydrous form of the drug may be substituted for the hydrate, but be sure to make appropriate dose corrections. For example, Sodium Sulfate USP is available as the decahydrate (Glauber’s salt) with a molecular weight of 322. The laxative dose of Sodium Sulfate decahydrate is 15 g. If the anhydrous form is substituted (MW = 142), only 6.6 g should be used:
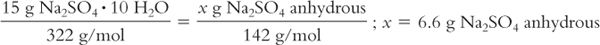
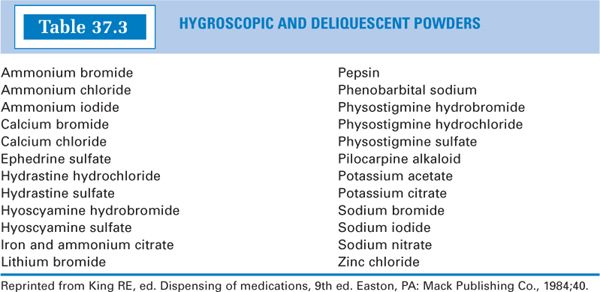
2. Hygroscopic and deliquescent drugs: Hygroscopic drugs or chemicals are solids that absorb moisture from the air. The term deliquescent refers to hygroscopic powders that may absorb sufficient moisture to dissolve and form a solution.
a. For examples of hygroscopic and deliquescent drugs, see Table 37.3.
b. Strategies for handling these drugs include the following:
(1) Store and dispense these drugs in tight containers. This means that powders dispensed as chartulae or divided powders should be sealed in plastic or foil and the packets put in tight containers. This is especially important in humid weather.
(2) For solid compounded formulations, an inert, powdered ingredient that will preferentially absorb water may be added to the formulation. Often there are sufficient other suitable powders in the formulation to fulfill this function. If not, a suitable, inert, water-insoluble (e.g., not lactose) powder may be added. Light magnesium oxide is sometimes used for this purpose and is acceptable, provided the quantity needed is not sufficient to impart a therapeutic laxative effect. When you add extra ingredients, consult with the prescriber. If necessary, make adjustments to maintain the intended dose.
(3) Counsel the patient to store the product or preparation in its original tight container and in a low-humidity environment.
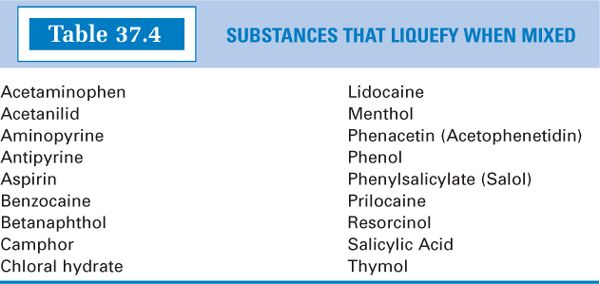
3. Pharmaceutical eutectic mixtures of drugs:A pharmaceutical eutectic mixture is defined as two or more substances that may liquefy when intimately mixed (as with trituration) at room temperature.
a. Under certain conditions, a “damp” powder, a pasty mass, or a liquid may result when two drugs or chemicals, which are solid at room temperature, are triturated together. This interesting phenomenon can easily be explained. It is well known that impurities present in chemicals impart melting points that are lower and less sharp than the melting point of the pure chemical. (Recall from organic chemistry lab that you checked for the purity of a compound by measuring its melting point; a clear, sharp melting point indicated pure compound.) If two or more drugs are triturated together, each may act as an impurity to the other and cause a mutual lowering of the original melting point of each individual compound. If the melting points of the pure compounds are low to begin with, the melting point of the mixture may be below room temperature, and a liquid or paste may result.
b. Whether liquefaction occurs or does not and the composition of the liquid or paste can both be analyzed using a phase diagram for the specific combination of compounds. For a more thorough discussion of this topic, consult a book of physical pharmacy (5,6). In general, liquefaction depends on the following:
(1) Ambient room temperature
(2) Original melting points of the substances
(3) Proportions in which the substances are mixed
(4) Extent and degree of pressure used in trituration
(5) Presence of other ingredients that may sorb any liquid formed
c. For examples of drugs that may form liquid eutectic mixtures, see Table 37.4.
d. There are some cases in which the formation of liquid eutectics is desirable.
(1) The local anesthetic EMLA Cream owes its success as a topical anesthetic, effective in preventing needle stick pain when drawing blood, to the high concentrations of lidocaine and prilocaine achieved by forming a liquid eutectic mixture of these two components.

(2) There are some solids that have hard crystalline structures that do not reduce to fine powder with direct trituration. An extra compounding step, known as pulverization by intervention (described in section V.B.2 of Chapter 25, Powders), is needed to make fine particles of this type of ingredient. However, if another ingredient in the formulation forms a liquid eutectic with this crystalline solid, the two can be triturated together, and the resulting liquid that is formed can either be adsorbed on an inert solid, as described later or, if the formulation is a dispersed system or a semisolid, the liquid can be directly incorporated in the formulation. This circumvents the need for the pulverization by intervention step. This method is illustrated with Sample Prescription 25.2 and is demonstrated on the CD that accompanies this book.
e. Strategies for handling drugs that form liquid eutectic mixtures include the following:
(1) Force the liquid eutectic to form, then sorb the liquid onto an inert, high-melting, finely divided solid. This is done by triturating together the eutectic forming drugs to force the formation of the liquid; then an inert powder is added in portions with trituration to sorb the liquid. (As mentioned earlier, this method is illustrated with Sample Prescription 25.2.)
(a) If possible, an ingredient already present in the formulation should be used to sorb the liquid.
(b) If no suitable powder is in the formulation, an inert powder may be added. Magnesium carbonate is reported to be the agent of choice, but light or heavy magnesium oxide, calcium phosphate, starch, talc, and lactose may also be suitable (5,7).
(2) An alternative method is to separately triturate each potential eutectic former with an inert powder, such as one of those given earlier; then the protected powders are mixed together by gentle spatulation.
(3) When you add extra ingredients, consult with the prescriber. If necessary, make adjustments to maintain the intended dose.
C. Changes in crystal form
1. Solid crystals are the most commonly encountered phase in pharmaceutical practice. Most pharmaceutical materials, either the active pharmaceutical ingredients or excipients, are in solid phase.
2. The solid phase can be further classified into two major types of subphases based upon the order of molecular packing. Crystalline, in which the molecules aggregate together with both short-range and long-range orders, is probably the most well known. The amorphous form, or glass, conversely, only presents short-range order but no long-range order in the way of molecular packing.
3. There are crystalline drugs that can exist in different crystalline structures in the solid state, although they are identical in the liquid or vapor state. Different polymorphic forms of the same substance will exhibit different physical properties, such as melting points and rates of dissolution.
a. Examples include ampicillin, methylprednisolone, hydrocortisone, various sulfa drugs, and barbiturates.
b. Because different polymorphs can give different dissolution rates, the use of different polymorphic forms in a solid dosage form can affect the drug’s bioavailability. This is a problem that has long been recognized by the pharmaceutical industry and is one of the reasons behind bioequivalence testing to ensure equivalent therapeutic performance of solid dosage forms made by different manufacturers.
c. The pharmacist should realize that metastable polymorphs are sometimes used in manufacturing solid dosage forms to give more rapid dissolution and improved bioavailability. Manipulation of these products in compounding may result in reversion to the more stable, less soluble, less available polymorph.
d. One common pharmaceutical substance that is notorious because of the problems caused by its polymorphic reversions is cocoa butter. Cocoa butter has several polymorphic forms, with melting points of 18°, 24°, 28° to 31°, and 34°C. Cocoa butter is used as a base for making suppositories and must be melted when the suppositories are made by fusion. It can very easily be overheated, and when it is, it solidifies as one of the lower melting polymorphs, which may melt at room temperature, or the suppositories may liquefy when handled by the patient during insertion. To avoid this problem, cocoa butter must be melted slowly and carefully, with the temperature not exceeding 34°C. This is illustrated with Example 31.1 in Chapter 31, Suppositories.
4. Pseudopolymorphs, such as hydrates, solvates, and co-crystals, may also form when drug powder are exposed to water, solvent, or other excipients during processing. The situation with hydrates or solvates can be more complex. Compared to solvates and co-crystals, hydrates play a much more important role in drug development. A survey conducted in 1999 indicated that more than 90 hydrates were included in USP monographs (8). A similar survey done on compounds in the European Pharmacopoeia indicated that approximately 29% of 808 organic compounds can form hydrates (9).
5. The amorphous state, also called glass, has many important applications, and lots of intensive studies have been devoted to these solids. However, the structural elucidation of amorphous materials is still incomplete. Glasses are formed by temperature quenching, fast evaporation of solvents, lyophilization, vapor condensation, mechanical stress, and the like. Although these pathways are different, they all kinetically avoid crystallization (except for mechanical stress) and keep the molecules in the coordinations as they were in liquid state. Amorphous solids are thermodynamically less stable than are crystalline solids.
D. Precipitation from solution
1. General principles
a. As stated at the beginning of this chapter, unintended precipitation of an active ingredient or excipient from solution can be a major hazard with pharmaceutical solutions.
(1) For oral or topical solutions, if an active ingredient precipitates, the particles will usually settle to the bottom of the container so that the initial doses poured from it will be subpotent and the later doses will be superpotent. This can result in either therapeutic failure or toxicity.
(2) With intravenous solutions, the danger from precipitation can be even greater because insoluble particles can lodge in capillaries and block them, resulting in severe consequences and even death.
b. Factors that can cause precipitation are discussed later, but it should be realized that pharmaceutical solutions are usually complex, and prediction of precipitation in them is never simple. Numerous reports in journal articles have pointed out that when reading and interpreting compatibility studies involving precipitation, all conditions of the study are relevant and may affect the outcome (10).
c. When interpreting compatibility reports, it is important to note: (i) the manufacturers of the drugs, (ii) the drug concentrations, (iii) the base solution or diluents and their manufacturers, (iv) order of mixing, (v) time frames, (vi) temperature, and (vii) test methods. Changes in any of these factors can alter the results. Two examples:
(1) The combination of injectable solutions of dopamine hydrochloride (12.8 mg/mL) and furosemide (5 mg/mL) had been reported to be compatible when mixed in usual IV solutions. It was later noticed that combining these same drugs in a Y-site administration gave a precipitate. It was subsequently learned that with this particular Y-site administration, one of the drug solutions was from a different manufacturer that used a different buffer, and a different pH resulted (10).
(2) In a study testing the physical compatibility of a large number of pairs of drug solutions, it was found that while some combinations were always compatible and some combinations were always incompatible, there were some cases for which compatibility depended on the order of mixing the two drug solutions; for some, the length of time after mixing was a factor (initial, 1-hour, and 3-hour testing was done) (11).
2. Solvent effects: When a drug is dissolved in a solvent and a second solvent, one in which the drug is poorly soluble, is added, the drug may precipitate.
a. Typical examples of solvent effects:
Topical: When water or an aqueous solution is added to an alcoholic solution of salicylic acid, precipitation may occur. (The solubility of salicylic acid is 1 g/2.7 mL of alcohol but only 1 g/460 mL water.)
Oral:When an aqueous solution or syrup is added to Phenobarbital Elixir, precipitation may occur. See Example 37.6 for information on the solubility of phenobarbital in various solvents. Injectables: Digoxin has a water solubility of 0.08 mg/mL. Digoxin Injection is available in a cosolvent system containing 40% propylene glycol and 10% alcohol. If it is diluted with an aqueous injectable solution, precipitation may occur.
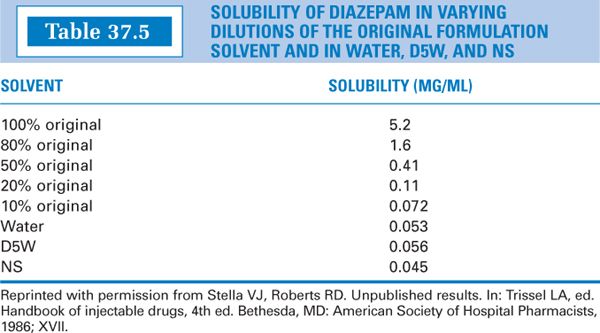
Another drug with similar problems is diazepam. Diazepam Injection has the following formula (12):
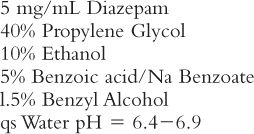
Observe the data in Table 37.5 and note the following results if Diazepam Injection is diluted with an aqueous injection such as Dextrose 5% in Water (D5W). In each case, 1 mL of Diazepam Injection (that is, 5 mg of the drug) is used.
Dilute 50:50: 1 mL Diazepam Injection and 1 mL D5W (that is, 50% original), gives 5 mg/2 mL = 2.5 mg/mL, which is >0.41 mg/mL; result, precipitation.
Dilute 1:10: 1 mL Diazepam Injection and 9 mL D5W (that is, 10% original) gives 5 mg/10 mL = 0.5 mg/mL, which is >0.072 mg/mL; result, precipitation.
Dilute 1:100: 1 mL Diazepam Injection and 99 mL D5W: 5 mg/100 mL = 0.05 mg/mL, which is approximately equal to 0.056 mg/mL; result, just at the borderline.
b. Usually the problem occurs when water is added to an alcoholic solution of a drug that is poorly soluble in water. The reverse can also be true. For example, codeine phosphate is very soluble in water, but it is not very soluble in alcohol. If alcohol is added to an aqueous solution of codeine phosphate, the drug may precipitate. This is why cough syrups that contain a large percentage of alcohol to solubilize other water-insoluble ingredients contain codeine base rather than codeine phosphate.
c. Remember that the useful (though approximate) relationship between solubility and solvent volume-fraction is logarithmic, not linear:
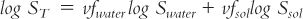
where:
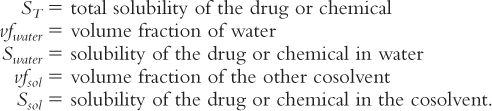
An acetaminophen solution is desired with a concentration of 325 mg/5 mL. The pharmacist finds the following solubility information for acetaminophen in Remington:The Science and Practice of Pharmacy: 1 g/70 mL water; 1 g/10 mL alcohol (13).
1. Express the total desired solubility (ST) and the solubility in water (Swater) and alcohol (Salc) in common terms (e.g., mg/mL):
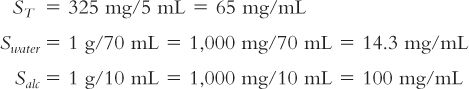
2. Recall that the sum of volume fractions = 1
Therefore, vfwater + vfalc = 1; and vfalc = 1 − vfwater
3. Substituting in the log solubility equation just given and solving for vf water:
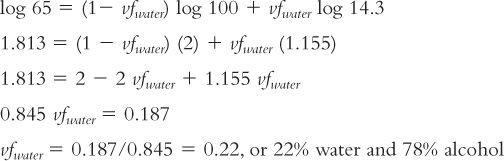
Remember that this equation is for pure systems containing the drug and the cosolvent system and that the result is just a good estimate. In formulation situations, we usually have other ingredients present, such as sweeteners, flavors, and other active ingredients and excipients. These factors alter the conditions and the results. The use of this equation is illustrated in Sample Prescription 27.5 in Chapter 27, Solutions.
d. Compounding strategies when adding additional solvents to a drug solution
(1) To maintain a true solution, be sure you are using an appropriate solvent system.
(a) For solubilities, consult appropriate references, such as Remington: The Science and Practice of Pharmacy or The Merck Index. If necessary, you may calculate approximate required solvent ratios using the log solubility equation given earlier.
(b) If you need to make an aqueous dilution of an injectable drug solution that contains a drug in a cosolvent system, follow the manufacturer’s instructions in the product package insert or recommendations in a reference, such as Trissel’s Handbook of Injectable Drugs. For example, a manufacturer of Digoxin Injection recommends that it be diluted with at least a fourfold volume of Sterile Water for Injection or the equivalent.
(c) If information is not available, make a reasonable dilution and observe the solution for a period of time, or dilute the product so that the final concentration of the drug is below its saturation concentration. In all cases, observe the solution and be sure to give sufficient time for redissolution if precipitation occurs.
(d) Decrease the drug concentration so that the drug is soluble in the solvent system. For systemic medications, remember that if you change the concentration of the active ingredient(s) in the preparation, you must change the volume of the dose administered to give the same quantity of drug.
(2) For oral or topical products, you may wish to make a suspension. A suspending agent may be required. Remember that a “Shake Well” label is required for dispersed systems.
(3) If you make a substantive change, you should first consult the prescriber.
3. pH Effects:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


