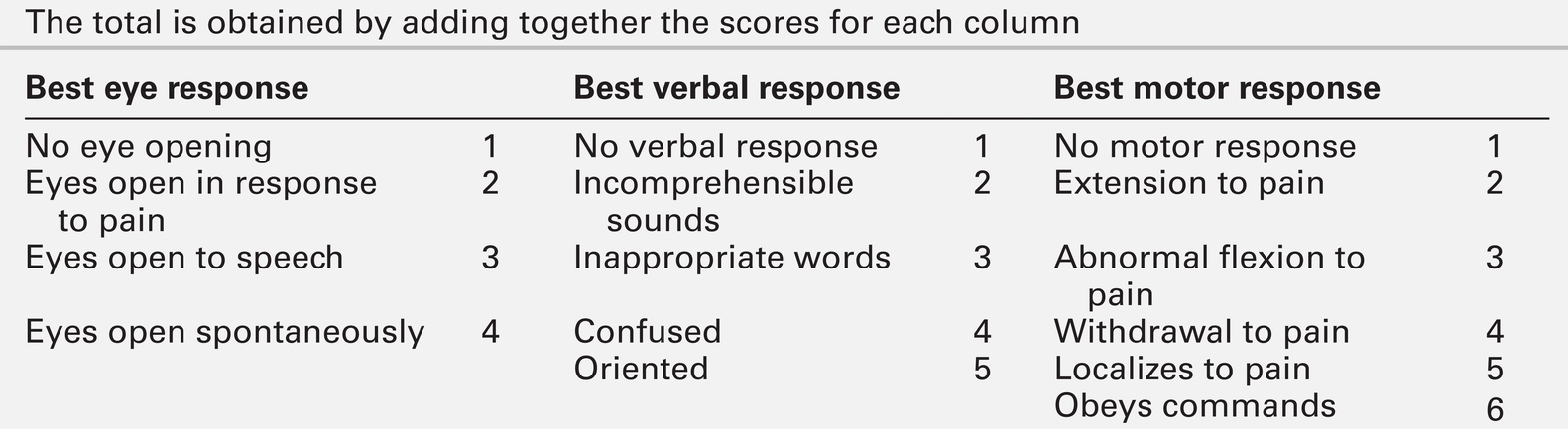
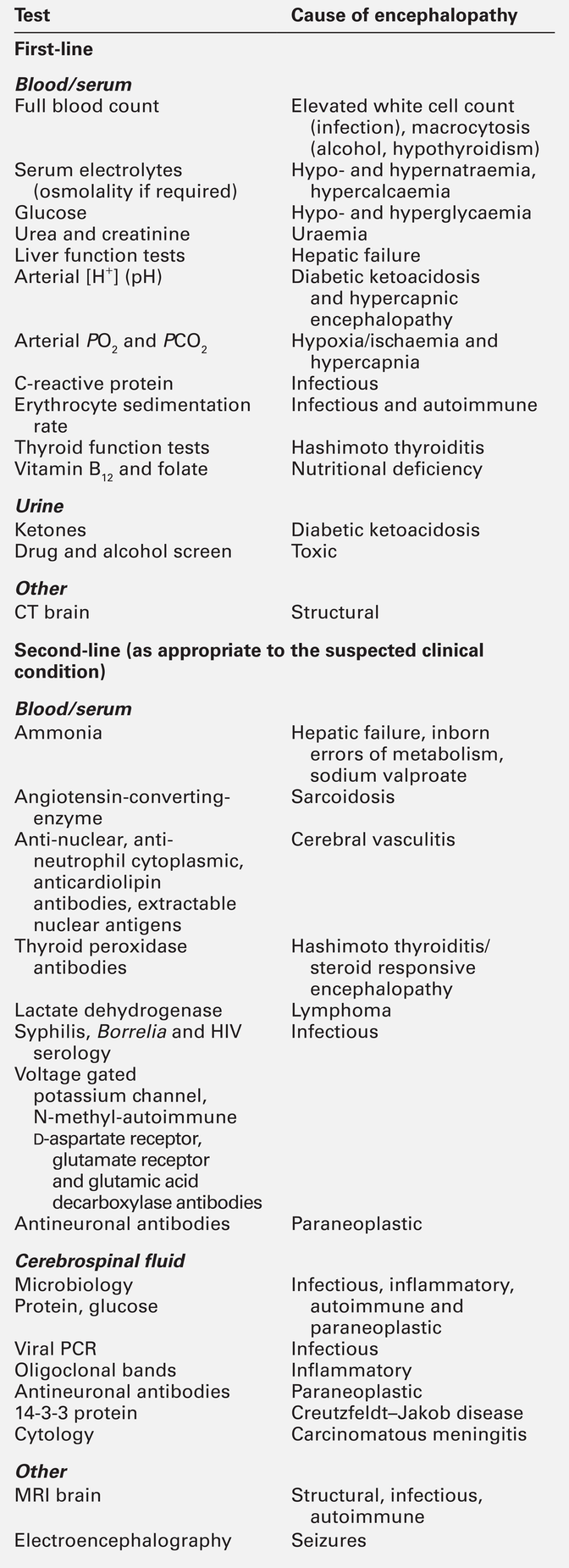
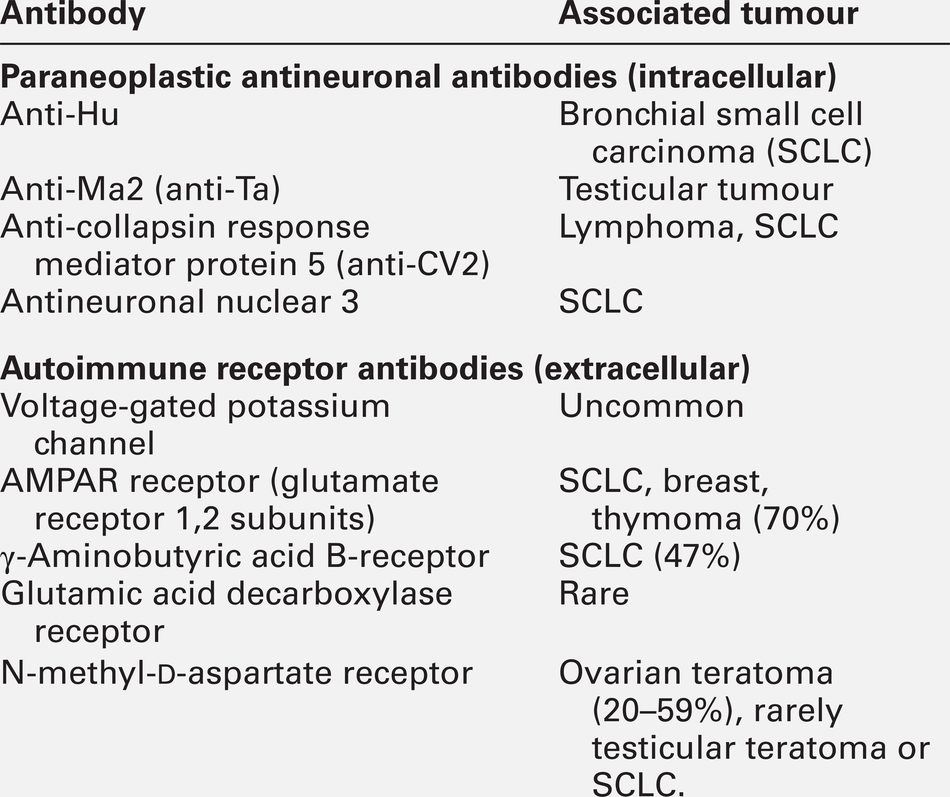
CHAPTER 36 Paul Hart; Clare M. Galtrey; Dominic C. Paviour; Min Htut CHAPTER OUTLINE Toxic and metabolic encephalopathy Vitamin B12 deficiency (subacute combined degeneration of the spinal cord) Small fibre painful axonal neuropathy Acute inflammatory neuropathies and variants Chronic kidney disease and established renal failure Nutritional peripheral neuropathies Neuropathy associated with bariatric surgery Ataxia with isolated vitamin E deficiency Early onset ataxia with oculomotor apraxia and hypoalbuminemia Fragile X-associated tremor/ataxia syndrome Hexosaminidase deficiency (GM2 gangliosidoses) Cerebrotendinous xanthomatosis (cholestanolosis) Neuronal ceroid lipofuscinosis INFLAMMATORY DISORDERS OF THE CENTRAL NERVOUS SYSTEM Despite the significant advances seen in laboratory and imaging diagnostics, many of the conditions seen by neurologists are still diagnosed on purely clinical grounds. In neurology, there remains no substitute for a meticulous clinical history and careful examination. Neurological investigations include all imaging modalities, neurophysiological tests of both the central and peripheral nervous system, and laboratory based investigations, including haematology, biochemistry, immunology, histopathology and cytology. This chapter will focus on the neurological conditions for which biochemical abnormalities are pertinent. Separate chapters cover the biochemistry of cerebrospinal fluid and muscle disease and other conditions that overlap with general neurological practice. ‘Encephalopathy’ is a term for any diffuse disease of the brain that alters brain function or structure. It is a common condition that encompasses coma, acute confusional state, delirium and dementia. The hallmark of encephalopathy is an altered mental state. Encephalopathy is caused either by processes that directly affect the structure of the brain or by systemic and metabolic factors (see Box 36.1). Encephalopathy can occur acutely (over hours to days); subacutely (over weeks to months) or chronically (over years) (Table 36.1). Although the mechanism of many causes of encephalopathy is well understood, the pathophysiology of metabolic encephalopathy is less clear but is likely to involve changes in amino acids and neurotransmitter profile. For patients presenting with altered consciousness, a detailed history often has to be taken from family, friends and other healthcare professionals, to determine pre-existing medical conditions and time of onset of altered mental state. For example, patients presenting with altered consciousness following apnoea or cardiac arrest are likely to be suffering from a hypoxic–ischaemic encephalopathy but alternative causes need to be excluded. Access to medication, previous psychiatric problems or use of illicit drugs may suggest intoxication. If the patient is known to have diabetes, hypo- or hyperglycaemia is likely. Patients on diuretics are at risk of hyponatraemia. Both a general systemic examination and detailed neurological examination are required to determine the cause of encephalopathy. Specific breath smells suggest certain causes, e.g. ammonia-like (uraemia); fruity-smelling (ketoacidosis); musty or fishy (acute hepatic failure); onion (paraldehyde), and garlic (organophosphates). Hypertension may indicate amfetamine or cocaine intoxication or a hypertensive encephalopathy. Examination of the skin may show signs of chronic liver disease, haemodialysis fistula, previous sternotomy scars from cardiac surgery or injection marks from intravenous drug use. Neurological examination should be divided into focal (localizing) and global (non-localizing) signs. Global signs suggest a process affecting the whole brain and include altered consciousness (acute confusional state, delirium and coma). The level of coma can be quantified with the Glasgow Coma Score, for which the minimum score is 3/15 (Table 36.2). TABLE 36.2 Glasgow coma scale (GCS) Other global neurological signs include generalized seizures, tremor, asterixis (abnormal jerking tremor in hands seen in liver flap) and myoclonus (involuntary brief jerk-like twitching, often multifocal). Focal signs indicate a localized problem within one part of the brain, for example visual or language disturbance from cerebral cortex pathology or eye movement disturbance, slurred speech or swallowing problems from brainstem pathology. Focal neurological signs usually suggest a primary neurological cause of encephalopathy. Two-thirds of encephalopathies are secondary to systemic metabolic factors. It is therefore important that these are excluded before assuming there is a neurological cause. Many metabolic encephalopathies are reversible if corrected promptly. Investigations for a patient with encephalopathy should include blood tests, urinalysis, imaging studies, and, when indicated, cerebrospinal fluid examination and an electroencephalogram. Increasingly, laboratory investigations can be used to make a definite diagnosis of neurological causes of encephalopathy owing to the improved recognition of antibodies for autoimmune encephalitis syndromes and biomarkers in neurodegenerative disease (Table 36.3). Toxic encephalopathy is caused by exogenous substances including solvents, drugs, radiation, paints, industrial chemicals, and certain metals. Some of the more common causes are discussed below. More details can be found in Chapter 40. This colourless and odourless gas is the most common cause of accidental poisoning in Europe and North America, as well as being a major cause of suicidal deaths. When 20–30% of total haemoglobin is bound to carbon monoxide, it causes headache, nausea and shortness of breath; when 50–60% is bound, coma results. Carbon monoxide exposure is detected by measuring the carboxyhaemoglobin concentration in blood. Carbon monoxide poisoning is treated with oxygen, using hyperbaric facilities in severe cases, if available. Alcohols, including ethanol, produce an altered mental state by central nervous system depression. Methanol and ethylene glycol similarly cause direct central nervous system depression but are also toxic via metabolism in the liver to formic acid (methanol) and glycolic and oxalic acids (ethylene glycol). Methanol and ethylene glycol concentrations can be measured in the blood but usually the suspicion is raised by other biochemical abnormalities. Poisoning with either causes a high anion gap metabolic acidosis. Treatment is by blocking the action of alcohol dehydrodenase with ethanol or fomepizole to decrease the production of toxic metabolites. Overdose causes coma, pinpoint pupils and respiratory depression. This can be from iatrogenic, accidental or suicidal overdose (e.g. morphine) or from use of illicit drugs (e.g. heroin). Diagnosis is usually suspected from the clinical triad but a rapid urine screening test can confirm opioid exposure. Treatment is with naloxone, given as a bolus dose followed by an infusion if the patient responds, in addition to supportive care. Wernicke–Korsakoff encephalopathy is a clinical triad of confusion, ataxia and ophthalmoplegia (eye movement disorder). It comprises two syndromes. First, Wernicke encephalopathy, an acute or subacute confusional state with ataxia and ophthalmoplegia and second, Korsakoff dementia, characterized by severe amnesia and confabulation. The former is often reversible but the latter is irreversible and persistent. Wernicke–Korsakoff encephalopathy is caused by thiamin deficiency and is most commonly seen in alcoholics but can also occur in patients with anorexia, hyperemesis gravidarum, those receiving total parenteral nutrition, or in the context of malabsorption and refeeding syndrome. The role of measurement of plasma thiamin concentrations in diagnosis and monitoring remains controversial and functional assessment by assay of red cell transketolase is no longer used by most laboratories for diagnosis. Therefore, laboratory tests are aimed at excluding other conditions and providing evidence of risk factors (e.g. macrocytosis and deranged liver function tests in alcoholism). Brain magnetic resonance imaging (MRI) can show signal change in the thalami, mammillary bodies, tectal plate and periaqueductal grey matter. Treatment is with parenteral thiamin. In the UK, normal practice is to give thiamin in a vitamin complex of riboflavin, nicotinamide, pyridoxine and ascorbic acid. Early treatment can reverse the symptoms rapidly and prevent dementia. Administration of intravenous glucose (including as part of parenteral nutrition) to patients who are severely malnourished, can exhaust their (already depleted) supply of thiamin and precipitate Wernicke–Korsakoff encephalopathy. Thiamin should therefore be administered before starting a glucose infusion in patients at high risk. This causes a typical pattern of degeneration of the white matter producing encephalopathy, myelopathy (subacute combined degeneration of the spinal cord), peripheral neuropathy and optic neuropathy. Vitamin B12 concentrations are easily measured and, if deficiency is corrected early, further damage can be prevented (See p. 688 and p. 693). Hepatic encephalopathy occurs as a consequence of liver function disturbance and its severity is graded from 0 to 4 (Table 36.4). TABLE 36.4 Grading of hepatic encephalopathy (West Haven classification system) Two situations can lead to severe hepatic encephalopathy – acute fulminant hepatic failure and decompensation of chronic liver disease. In the UK, acute liver failure is most commonly from paracetamol overdose or viral hepatitis, and chronic liver disease from alcohol abuse or chronic hepatitis B or C infection. Encephalopathy is thought to develop because hepatocellular dysfunction produces neurotoxins (e.g. ammonia), false neurotransmitters and benzodiazepine-like substances. In chronic liver disease, there can be the additional effect of porto–systemic shunting. This allows significant quantities of ammonia, formed in the bowel from protein, to reach the systemic circulation. The severity of the neurological disorder correlates poorly with ammonia concentrations so measurement rarely contributes to management. ‘Liver function tests’ will confirm that the concentration of bilirubin is high, often with abnormal liver enzyme activities and, more importantly, poor synthetic function with abnormal clotting. Plasma paracetamol concentration can be measured if overdose is a possibility. Patients with acute liver failure may require liver transplantation for survival but the encephalopathy of chronic disease may be reversed by conservative measures (see Chapter 14 for further details). Uraemic encephalopathy presents with apathy, fatigue, inattentiveness and irritability, followed by confusion, hallucinations, slurred speech, tremor and asterixis. The mechanism is unclear but correction of uraemia reverses the encephalopathy. Suggested mechanisms include retention of organic acids, elevation of phosphate concentration in the cerebrospinal fluid (CSF), increased calcium content of the cerebral cortex owing to the action of PTH, and alteration of concentrations of neurotransmitters or proinflammatory cytokines. Although the degree of the rise in urea correlates with the severity of encephalopathy, it is not thought to be causative. The investigation, monitoring and treatment of uraemia are discussed in Chapter 7. Hypercapnia occurs in respiratory failure either secondary to lung disease (e.g. chronic obstructive pulmonary disease) or to mechanical problems such as neurological disease (e.g. myasthenia gravis). Clinically, hypercapnia presents with headache, papilloedema, mental slowing, drowsiness, confusion, coma and asterixis. The mechanism is unclear but thought to be due to a direct effect of carbon dioxide possibly on the hydrogen ion concentration (pH) of the CSF. Hypercapnia can be confirmed by measurement of PCO2 on an arterial blood sample. Coma can occur with PCO2 > 9 kPa. Treatment is of the underlying cause and, once corrected, there is no prolonged cerebral damage (see Chapter 5 for further details). Hypoxic–ischaemic encephalopathy is caused by hypoxaemia and/or reduced blood flow to the brain from failure of the cardiac system, the respiratory system or both (e.g. myocardial infarction, drowning). When cerebral perfusion pressure falls, there is a failure of autoregulation leading to ischaemia and hypoxia and to cell death by both necrosis and apoptosis. The diagnosis is usually evident from the history. Myoclonus is common (Lance–Adams syndrome). There is a wide spectrum of clinical outcomes depending on the degree and duration of the insult from full recovery to severe prolonged disability and death. Hypoglycaemia causes confusion, seizures and coma and can cause permanent neurological damage if not reversed quickly. Up to 15% of patients with diabetes will have at least one episode of hypoglycaemic coma in their lifetime. Recurrent hypoglycaemia, such as that caused by an insulinoma, can be mistaken for epilepsy. The brain contains 1–2 g of glucose stored as glycogen, which is utilized within approximately 30 min if blood glucose concentration remains low. Ketones can also be used as an energy source but this is not sufficient in prolonged hypoglycaemia, nor are they available when hypoglycaemia is caused by high insulin concentrations. The most common causes of hypoglycaemia are accidental or deliberate overdose of insulin, insulinoma, and depletion of liver glycogen (e.g. acute liver failure, alcohol binge, severe starvation). Hypoglycaemia is easily detected by blood glucose measurement at the bedside (although it should be confirmed by laboratory measurements) (see Chapter 17 for further details). Hyperglycaemic coma occurs in two forms – diabetic ketoacidosis and the hyperosmolar hyperglycaemic state. Coma is a late feature of diabetic ketoacidosis and develops when hyperglycaemia, dehydration, acidosis and shock are severe. Cerebral oedema is a rare but often fatal complication both of the condition and its treatment. The coma of the hyperosmolar hyperglycaemic state usually develops more insidiously and occurs in elderly patients, often with previously undiagnosed diabetes mellitus. Extreme hyperglycaemia with greatly increased plasma osmolality, electrolyte losses and dehydration exacerbated by impaired thirst awareness, are all thought to contribute to the development of the encephalopathy (see Chapter 16 for further details). Hyponatremia has many causes, including liver and cardiac failure, syndrome of inappropriate antidiuresis (neurological causes of which include head trauma, bacterial meningitis, encephalitis, cerebral infarction, subdural and subarachnoid haemorrhage), vomiting and diuretic use. The severity of the symptoms is related to the rapidity of decline in plasma sodium. This is because hyponatremia causes extracellular hyposmolarity and a tendency for free water to shift from the vascular space to the intracellular space. When plasma sodium concentration falls slowly, over a period of several days or weeks, the brain is capable of compensating by extrusion of solutes to the extracellular space. This reduces the flow of free water into the intracellular space and symptoms are much milder for a given degree of hyponatraemia. When the plasma sodium concentration falls rapidly, this compensatory mechanism is overwhelmed and severe cerebral oedema may ensue. For example, a sodium concentration falling from normal to < 110 mmol/L over 24–48 h will often cause coma. It is necessary to correct the plasma sodium concentration in a controlled manner because of the risk of central pontine myelinolysis if the increase is too rapid (see Chapter 4). Central pontine myelinolysis is characterized by quadriparesis, dysphagia, dysarthria, diplopia and altered consciousness. Areas of the brain other than the pons are also often involved. Magnetic resonance imaging reveals signal change within these areas but the appearances may be normal early in the course of the disease. Central pontine myelinolysis may prove fatal, and incomplete recovery is common in survivors. Common causes of hypernatraemia include diabetes insipidus, hyperglycaemia, diarrhoea and fluid deprivation. Again, it is the rate of change of sodium concentration that is important rather than the actual values. A sodium concentration of > 160 mmol/L can cause coma due to cerebral cellular dehydration if it develops rapidly. Treatment depends on the cause of the disorder and the total body fluid volume (see Chapter 4). Severe hypercalcaemia (> 3.4 mmol/L) can be lead to coma, preceded by fatigue, headache, muscle weakness, irritability and confusion. High concentrations of calcium ions decrease neuronal excitability, which together with dehydration is thought to cause the neurological problems (see Chapter 6). Severe extracranial infection can be associated with an impaired mental state. The pathophysiological mechanism involves reduced cerebral blood flow and oxygen extraction by the brain, cerebral oedema, disruption of the blood–brain barrier owing to inflammatory mediators acting on the cerebrovascular endothelium, and abnormal neurotransmitter compositions. Diagnosis of sepsis can be aided by the finding of a raised white blood cell count and raised inflammatory markers, and by blood cultures. Treatment is of the underlying infection with supportive care. The last decade has seen significant advances in our understanding of antibody-mediated encephalopathies. There are two types of antibodies detectable in blood in these conditions (Table 36.5): paraneoplastic antibodies to intracellular targets that are associated with cancer but are not pathogenetic, and antibodies to the extracellular domain of neuronal cell-surface proteins that directly cause encephalitis and are also often associated with cancer. Autoimmune encephalitis typically presents with subacute memory loss, psychiatric and behavioural disturbance and seizures. The detection of one of the specific antibodies supports a diagnosis of paraneoplastic encephalitis. Treatment is of the underlying cancer and with immunotherapy. Hashimoto encephalopathy presents with seizures, behavioural and psychiatric manifestations, movement disorders and coma and is associated with the presence of high titres of anti-thyroglobulin or anti-thyroid peroxidase (TPO, antimicrosomal) antibodies. The condition can present without any features of thyroid disease. It is unclear whether the anti-thyroid antibodies are pathogenetic or simply represent an immune epiphenomenon but it is important not to miss this diagnosis as the condition responds to treatment with steroids. Dementia is a syndrome characterized by progressive deterioration of cognitive function without alteration of consciousness. Cognitive deficits most commonly affect memory, but other cognitive domains such as language, praxis, visual perception and most notably executive function are also often affected. Diagnosis of dementia is generally by clinical criteria during life, although many of its causes are defined on the basis of histopathological criteria with the consequence that definitive diagnosis can only be made at post-mortem. However, over the last few years a number of laboratory CSF biomarkers have emerged and have an increasing role in diagnosis. Cerebrospinal fluid Aβ-40, Aβ-42, total tau and phosphorylated tau proteins are the most sensitive biomarkers for the diagnosis of Alzheimer disease and 14-3-3 protein for the diagnosis of Creutzfeldt–Jakob disease (see Chapter 34). Spinal cord disease usually presents with motor and/or sensory symptoms in either the upper and lower or just the lower limbs, depending on the site of the insult. Clinical signs include increased tone in the limbs, brisk reflexes and, typically, well-preserved muscle bulk. Bowel and bladder function can be compromised. The list of possible causes of spinal cord disease is extensive and includes vascular, degenerative, compressive, inflammatory, malignant, metabolic and infectious conditions. Metabolic causes include vitamin B12 deficiency (Box 36.2) and adrenomyeloneuropathy. A normal MRI scan of the spinal axis will exclude a significant number of differential diagnoses. Deficiency of vitamin B12 can produce haematological and neurological abnormalities: features of gastrointestinal disease may also be apparent (see Chapter 12). From an early stage, it is the spinal cord that is predominantly affected. The onset of symptoms is insidious, with symmetrical, uncomfortable, tingling paraesthesiae, initially in the feet but later involving the hands. As the condition progresses, the gait becomes ataxic (sensory ataxia owing to loss of proprioception) and the limbs become weaker and increasingly spastic. Additional problems include peripheral polyneuropathy, optic neuropathy, psychiatric disturbance and dementia. The exact pathophysiological process leading to neuronal damage in vitamin B12 deficiency is unknown. Vitamin B12 is involved as a cofactor in the conversion of homocysteine to methionine and of methylmalonyl- CoA to succinyl-CoA. It is possible that the impaired synthesis of methionine leads to a depletion of S-adenosylmethionine, which is required for myelin synthesis. The diagnosis is confirmed by measuring vitamin B12 concentration in the serum. In some patients, the concentration may be only ‘low-normal’. In borderline cases, measurement of homocysteine and methylmalonate concentrations can be useful. It is important that the diagnosis is made as early as possible as this is a potentially reversible condition. Magnetic resonance imaging of the spine often shows the spinal cord to be atrophic and can show signal change in the dorsal columns. Folate deficiency is usually associated with other nutritional deficiencies. Folate antagonists (e.g. methotrexate, trimethoprim and pyrimethamine) are also potential causes. Neurological manifestations are rare and the same as with vitamin B12 deficiency. Treatment is by folate replacement, although any vitamin B12 deficiency should be treated first to prevent exacerbation of spinal cord degeneration. Copper deficiency is rare and causes both haematological and neurological disease. Neurological manifestations include myelopathy, peripheral neuropathy and optic neuropathy. Copper deficiency was recognized in ruminants (swayback disease) long before it was identified in humans. The presentation can be similar to that of subacute combined degeneration of the spinal cord. It is usually secondary to gastric surgery, gastrointestinal diseases or total parenteral nutrition, or to excess intake of zinc or iron, which reduce copper absorption in the gut. Copper deficiency myelopathy has been attributed to ingestion of zinc contained in denture cream in some patients. Treatment is with oral or intravenous copper replacement. Vitamin E deficiency is rare, and causes a spinocerebellar ataxia, retinopathy, myopathy and anaemia. Poor diet is not usually sufficient cause unless associated with malabsorption. Abetalipoproteinemia is a rare inherited disorder of fat metabolism that results in poor absorption of dietary fat and vitamin E. Vitamin E deficiency is discussed further below (see ataxia). Hepatic myelopathy is a rare complication of chronic liver disease. It has been suggested that it is caused by the neurotoxicity of ammonia or other metabolites bypassing normal liver metabolism. It is characterized by spastic paraparesis with minimal sensory and sphincteric involvement. Diagnosis is a process of exclusion requiring normal CSF analysis and brain imaging. Liver transplantation may result in improvement, especially if performed early in the course of the disease. This is a progressive neurodegenerative disease with variable age of onset and rate of progression and hence variable prognosis. The clinical presentation of the chronic form of hexosaminidase A deficiency may mimic spinocerebellar degeneration, Friedreich ataxia, or amyotrophic lateral sclerosis (see below). This is the commonest clinical variant of adrenoleukodystrophy, a disorder of peroxisomal fatty acid oxidation causing the accumulation of very long chain fatty acids in myelin, adrenal cortex and Leydig cells of the testes. Patients with an adrenomyeloneuropathy presentation of this X-linked recessive disorder present in their third decade with a progressive spastic paraparesis and sphincter dysfunction. A sensorimotor peripheral neuropathy may be a feature and ataxia and dementia are seen occasionally. Adrenal insufficiency will often have been present since childhood and there may be hypogonadism. Around 50% of heterozygous females will show some symptoms later in life. The clinical findings and magnetic resonance imaging point to the diagnosis, which is confirmed by demonstrating hypoadrenalism and elevated concentrations of very long chain fatty acids (VLCFAs) in plasma and cultured skin fibroblasts. Genetic testing is available. The only gene in which mutations are known to cause adrenoleukodystrophy is ABCD1, which codes for a peroxisomal membrane transporter protein, although molecular genetic testing is rarely required to confirm the disease, especially in a male. Treatment is with steroid replacement, symptomatic management and supportive care.
Biochemical aspects of neurological disease
INTRODUCTION
ENCEPHALOPATHY
The total is obtained by adding together the scores for each column
Toxic and metabolic encephalopathy
Carbon monoxide
Alcohol
Opioids
Thiamin (vitamin B1) deficiency
Vitamin B12 deficiency
Liver failure
Grading
Symptoms
0
Minimal hepatic encephalopathy. Lack of detectable changes in personality or behaviour. Minimal changes in memory, concentration, intellectual function, and coordination. Asterixis absent
1
Trivial lack of awareness. Shortened attention span. Impaired addition or subtraction. Hypersomnia, insomnia, or inversion of sleep pattern. Euphoria, depression, or irritability. Mild confusion. Slowing of ability to perform mental tasks. Asterixis can be detected
2
Lethargy or apathy. Slurred speech. Obvious asterixis. Drowsiness, lethargy, gross deficits in ability to perform mental tasks. Obvious personality changes, inappropriate behaviour and intermittent disorientation, usually regarding time
3
Somnolent but can be aroused. Unable to perform mental tasks. Disorientation about time and place, marked confusion, amnesia. Occasional fits of rage, present but incomprehensible speech
4
Coma with or without response to painful stimuli
Chronic kidney disease and established renal failure
Respiratory failure
Cardiorespiratory failure
Disorders of glucose metabolism
Hyponatremia
Hypernatraemia
Hypercalaemia
Septic encephalopathy
Autoimmune encephalopathy
Dementia
SPINAL CORD DISORDERS
Vitamin B12 deficiency (subacute combined degeneration of the spinal cord)
Folate deficiency
Copper deficiency
Vitamin E deficiency
Hepatic myelopathy
Hexosaminidase A deficiency
Adrenomyeloneuropathy
PERIPHERAL NEUROPATHY

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


