OUTLINE
Training, Evaluation, and Responsibility of Compounding Personnel in Aseptic Manipulation Skills
CSP Microbial Contamination Risk Levels
Verification of Compounding Accuracy and Sterility
Environmental Quality and Control
Suggested Standard Operating Procedures
Verification of Automated Compounding Devices for Parenteral Nutrition Compounding
Finished Preparation Release Checks and Tests
Maintaining Sterility, Purity, and Stability of Dispensed and Distributed CSP
Patient Monitoring and Adverse Events Reporting
I. INTRODUCTION
A primary responsibility of the pharmacist is to ensure safe sterile dosage form preparation. Compounding an accurate formulation free of microbial and particulate matter is an essential component of this process. A number of procedures have been developed by such organizations as the USP and the American Society of Health-System Pharmacists (ASHP) to assist pharmacists in complying with sterile preparation admixture guidelines. As was described in Chapter 12 of this book, the USP is a private, nonprofit organization recognized by the federal government as the official group responsible for setting national standards for drug purity and safety. Most recently, the USP has become involved with issuing standards on the pharmaceutical compounding of sterile preparations. On January 1, 2004, the USP formally adopted Chapter 〈797〉, the first official chapter enforceable by regulatory agencies concerning the procedures and requirements for compounded sterile preparations (CSPs). Much of the same information was previously published as recommendations in nonenforceable materials including USP Chapter 〈1206〉, which focused on dispensing for home care, and ASHP Guidelines on Quality Assurance for Pharmacy-Prepared Sterile Products, which gave standards and procedures for sterile dosage form preparation in hospitals and institutional settings. Since 2005, Chapter 〈797〉 has undergone major revision and, in December 2007, a new and expanded document was posted on the USP’s Internet Web site. These revised standards take effect on June 1, 2008; they were published in the 2008 USP Pharmacists’ Pharmacopeia and are scheduled for publication in the 2009 USP 32/NF 27.
The following is a summary of guidelines necessary for accurate and safe preparation of CSPs. For a more complete discussion of this topic, refer to USP Chapter 〈797〉, which is now considered to be the standard of practice for this area of pharmaceutical compounding (1).
II. DEFINITIONS
A. Ante-area: an area adjacent to the clean room, which, although of high quality, may have a lesser air cleanliness classification than does the clean room. The ante-area should be maintained as an International Organization for Standardization (ISO) Class 8 or higher (see Table 32.1 for ISO classifications). Activities in the ante-area include such things as hand washing and gowning and unpacking of clean-room supply packages. Cardboard boxes and other packaging materials are not brought into clean rooms, because opening and handling them introduces particulates into the environment.
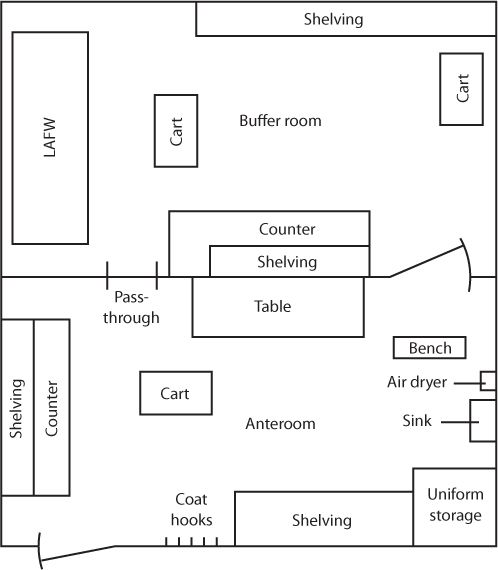
SAMPLE FLOOR PLAN FOR A CLEAN ROOM AND ANTE-AREA
B. Beyond-use date (BUD): the time and date after which a CSP should not be stored or transported. It is determined from the time and date the CSP is compounded. The BUD definition in USP Chapter 〈797〉 also references USP General Notices and Chapter 〈795〉 Pharmaceutical Compounding—Nonsterile Preparations. The General Notices defines BUD as, “the date after which a compounded preparation may not be used” (2) (See Chapter 4, Expiration and Beyond-Use Dating, of this book for additional information on this topic.)
C. Biological safety cabinet (BSC): a cabinet that provides an environment for aseptic preparation of CSPs. It has an open front with inward vertical airflow to protect the worker from contamination by hazardous drugs and a downward airflow blown through a high-efficiency particulate air (HEPA) filter for preparation and environmental protection.
D. Buffer area: ISO Class 7 environment that houses devices used in aseptic compounding. Such devices include, but are not limited to, laminar airflow workbenches (LAFWs), BSCs, or compounding aseptic isolators (CAIs).
E. Clean room: a room that is designed and maintained to meet a specified airborne particulate cleanliness class, such as Class 1,000 (ISO Class 6) or Class 10,000 (ISO Class 7). Clean rooms contain LAFWs in order to prevent particulate and microbiologic contamination of drug preparations as they are being prepared or processed.
F. Compounding aseptic containment isolator (CACI): a compounding aseptic isolator (CAI) designed to protect the worker from cytotoxic or hazardous airborne drug particles during the compounding or material transfer process. Air should first pass through a microbially retentive HEPA filter before exchange with the surrounding environment. The air exhaust from the isolator should be properly vented from the building when volatile drugs are processed within the isolator.
G. Compounding aseptic isolator (CAI): an isolator designed for the aseptic compounding and material transfer of pharmaceutical preparations. A HEPA filter should be used with a CAI for air exchange with the surrounding environment. These devices are sometimes referred to as barrier isolators.
H. Critical area: an ISO Class 5 environment where CSPs, containers, and closures are processed.
I. Critical site: any surface (e.g., vial septa, injection port) or opening (e.g., opened ampules, needle hubs) that is at risk for contamination through direct contact with air, moisture (e.g., oral secretions), or touch.
J. Closed-system vial transfer devices (CSTDs): vial transfer systems that allow no venting or exposure of hazardous substances into the environment.
K. Direct compounding area (DCA): the critical area within an ISO Class 5 primary engineering control (that is, an LAFW, BSC, CAI, etc.) where critical sites are exposed to unidirectional HEPAfiltered air, also know as first air.
L. Disinfectant: a chemical or physical agent used to eliminate harmful pathogens. These agents may not necessarily kill microorganisms or fungal spores.
M. Endotoxin: a pyrogenic product present in bacterial cell walls. These substances are lipopolysaccharides that can be found anywhere live or dead bacteria have been present. As large molecules, they are not destroyed by steam sterilization or bacterial filtration. They can be destroyed on glassware using dry heat sterilization. The most common source of endotoxin material is water that has contained bacterial contamination; the endotoxin can remain after any bacteria are removed or killed. When a solution containing the endotoxin in injected into a patient, it can cause fever and even death.

COMPOUNDING ASEPTIC ISOLATOR (Photo Courtesy of Containment Technologies Group, Inc.)
N. First air: refers to the air that has passed through a unidirectional HEPA filter that is free of contaminants.
O. HEPA filter: a filter that provides a HEPA (or high-efficiency particulate air) environment, which is an essential component of both horizontal and vertical laminar airflow workbenches and other aseptic processing areas. For these purposes, the HEPA filters are certified to provide air that is filtered with a minimum 0.3-μm particle retaining efficiency of 99.97%.
P. Laminar airflow workbench (LAFW): also known as laminar flow hoods, workbenches that provide an environment of specially filtered air that sweeps the work area and provides an aseptic work area. Regular room air is drawn through a gross filter into an intake opening in the hood; the air then goes through a plenum where the airflow is equalized and is then passed in a unidirectional parallel flow pattern (laminar flow) through a HEPA filter. The air is forced through the HEPA filter and over the work area at a velocity of 90 ft/min, which is sufficient to sweep particulate matter away from the work area.
With horizontal flow hoods, the HEPA filter takes up the back, vertical surface of the hood space, and the laminar air blows from the HEPA filter horizontally across the work area and directly at the worker who is standing at the front edge of the hood and working on the workbench surface.
With vertical flow hoods, the HEPA filter takes up the top, horizontal surface of the hood space, and the laminar air blows from the HEPA filter downward through the hood space and into intake grills located along the front and back edges of the workbench surface. Vertical flow hoods, also called biological safety cabinets (BSCs), can be used for any aseptic processing, but they are required for working with cytotoxic or other hazardous drugs. They have a clear glass or plastic shield that comes partway down the front of the hood space; the clear front shield and the vertical flow pattern with air contained within the hood protect the worker from contamination by drugs being processed within the hood.
Q. Media fill: a procedure in which personnel who do aseptic processing of CSPs prepare a simulated preparation using microbiologic growth medium, such as Soybean-Casein Digest Medium. The simulated preparation is then incubated to determine whether the preparation was contaminated during the procedure. The media-fill procedure attempts to simulate as closely as possible the exact environmental and process-related conditions and intensity level (e.g., time during the work shift and number of transfers or manipulations required to create a preparation) of actual practice; the verification procedure should be representative of the greatest risk that might be experienced in an actual practice situation.
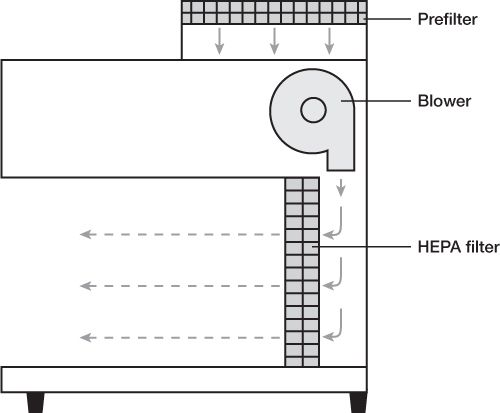
HORIZONTAL LAMINAR AIRFLOW WORKBENCH (LAFW)
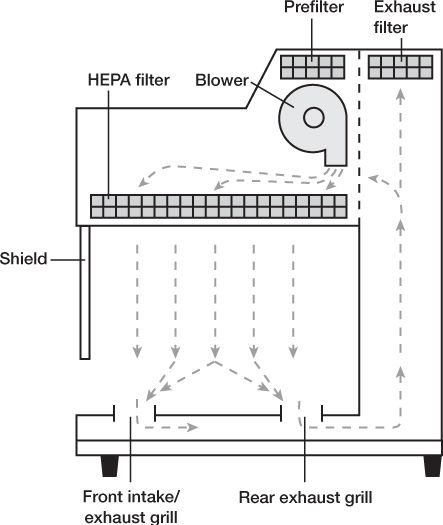
VERTICAL LAMINAR AIRFLOW WORKBENCH (LAFW)
R. Multiple-dose container: a container with antimicrobial preservatives consisting of several doses of a preparation that is intended to be used multiple times. A beyond-use date (BUD) of 28 days (based on the day the container is first opened or entered) is given for multiple-dose containers with antimicrobial preservatives unless a shorter date is specified by the manufacturer.
S. Negative-pressure room: a room that is at a lower pressure than the adjacent spaces so that the net flow of air is into the room (3).
T. Pharmacy bulk package: a container that holds many single doses of a sterile preparation intended for parenteral administration or the compounding of parenteral admixtures only. The closure of the pharmacy bulk package should be penetrated only once for transfer and should be manipulated only in a clean room. Pharmacy bulk packages must be labeled as “Pharmacy Bulk Package—Not for Direct Infusion,” contain information referring to the safe use of the preparation, and list detailed information about the time period in which the preparation may be used once it has been entered, given that it has been stored in accordance with the labeled conditions.
U. Primary engineering control (PEC): an ISO Class 5 device or room that provides the proper environment for preparation of CSPs. Such devices include, but are not limited to, LAFWs, BSCs, CAIs, and CACIs.
V. Preparation: a sterile or nonsterile pharmaceutical dosage form or nutrient that is compounded in a licensed pharmacy or healthcare-related facility in accordance with an order from a licensed prescriber.
W. Product: a drug, pharmaceutical dosage form, or nutrient that is commercially manufactured in an FDA-approved facility. Such a product has been evaluated by the FDA for safety and efficacy, and FDA-approved labeling and product information accompanies the product.
X. Positive-pressure room: a room that is at a higher pressure than adjacent spaces so that the net airflow is out of the room (3).
Y. Pyrogen: a substance that induces a fever in a patient.
Z. Single-dose container: a container holding a preparation intended for a single use and for parenteral administration only. One such example is a prefilled syringe.
AA. Segregated compounding area: a separate designation area (but not an ISO Class 7 environment) containing an ISO Class 5 PEC used for the preparation of low-risk level CSPs with a BUD of 12 hours or less.
BB. Sterility: the absence of viable microorganisms. Because sterility cannot usually be confirmed with absolute certainty, statistical probability is used to describe it.
CC. Sterilizing-grade membranes: filter membranes that “retain 100% of a culture of 107 microorganisms of a strain of Brevundimonas (Pseudomonas) diminuta per square centimeter of membrane surface under a pressure of not less than 30 psi (2.0 bar). Such filter membranes are nominally at 0.22-μm or 0.2- μm porosity” (1).
DD. Terminal sterilization: a lethal procedure that is carried out at the end of processing, when a product or preparation is in its final sealed container, with the purpose of achieving a sterility assurance level of less than 10–6 (that is, the probability of a nonsterile unit is greater than one in a million) (4).
EE. Unidirectional flow: air flowing in a single and uniform manner that sweeps the work area and provides an aseptic processing area.
III. TRAINING, EVALUATION, AND RESPONSIBILITY OF COMPOUNDING PERSONNEL IN ASEPTIC MANIPULATION SKILLS
A. “Compounding personnel are responsible for ensuring that CSPs are accurately identified, measured, diluted, and mixed and are correctly purified, sterilized, packaged, sealed, labeled, stored, dispensed, and distributed” (1).
B. Health care professionals who supervise compounding personnel shall ensure the following:
1. Personnel are adequately trained and educated.
2. Ingredients are correctly identified, in terms of quality and purity.
3. Open or partially used containers are properly stored.
4. Proper sterilization techniques are used for CSPs.
5. Compounding equipment and devices are kept clean and maintained as accurate.
6. CSPs are evaluated for potential harm from added substances prior to dispensing and administration.
7. Appropriate packaging for CSPs is used to maintain sterility and stability.
8. Compounding environments maintain purity or sterility of CSPs.
9. CSP labels are accurate and complete.
10. BUDs are valid based on scientific criteria or direct testing.
11. Compounding procedures are in accordance with established criteria.
12. Compounding deficiencies can be identified rapidly and corrected.
13. Compounding procedures are separate from quality review.
C. Personnel involved in the preparation of CSPs must be trained through didactic instruction in the theory of sterile compounding and by practical skills to perform aseptic manipulations.
D. Compounding personnel must pass written and media-fill testing initially and at least annually for low- and medium-risk level conditions and semiannually for high-risk level conditions.
E. Should compounding personnel fail written tests or produce gross microbial colonization within media-fill test vials, expert compounding personnel will immediately retrain and reevaluate these personnel to correct all deficiencies in aseptic practices.
F. Cleaning and disinfection of sterile compounding areas
1. Food, drinks, and other similar products must never be brought into areas where components and ingredients used for the preparation of CSPs are present. This includes ante-areas, buffer areas, and segregated compounding areas.
2. Surfaces of LAFWs, BSCs, CAIs, and CACIs must be cleaned and disinfected at the beginning of each work shift, prior to each batch preparation, every 30 minutes during prolonged periods of CSP preparation, and during heavy surface contamination such as a spill.
3. When no compounding of CSPs is occurring, floors of all ISO Class 7 and 8 areas, buffer areas, and ante-areas must be cleaned and disinfected by mopping.
4. Cleaning materials used in the compounding areas, such as wipers and sponges, must be nonshedding and remain in these areas until disposal.
5. If it is deemed acceptable to reuse specific cleaning materials (e.g., mops) based on manufacturer recommendations, procedures must be developed to ensure the efficacy of these materials while preventing contamination of the area on repeated use.
6. A suitable disinfecting agent [e.g., sterile isopropyl alcohol (IPA)] must be used to wipe clean compounding supplies and equipment when removed from shipping boxes.
7. Sterile IPA should remain on compounding surfaces at least 30 seconds to allow complete disinfection prior to initiating CSP preparation.
G. Personnel cleansing and garbing
1. Personnel must be properly trained about the use of protective equipment (i.e., gloves, gowns, head covers, facemasks) to ensure aseptic preparation of CSPs.
2. Personnel experiencing health problems that may increase the burden of airborne particulates must not be allowed to participate in aseptic compounding procedures. Examples of such health conditions include rashes, weeping sores, and active respiratory infections.
3. All personal outer garments (e.g., hats, sweaters), cosmetics, jewelry, body piercings, and artificial nails must be removed prior to entering the buffer or compounding areas as these materials can increase particulates into the air or interfere with the effectiveness of protective equipment.
4. Personnel garbing and cleansing must be performed in an order considered to progress from the dirtiest to the cleanest activity, such as placement of (i) shoe covers, (ii) hair covers and facemasks, (iii) eye shields, (iv) fingernail cleaning, (v) hand and forearm washing, (vi) nonshedding gown, and (vii) sterile gloves.
5. Prior to donning sterile gloves, personnel must clean their hands with an alcohol-based surgical scrub possessing persistent activity and wait until their hands are completely dry.
6. Gloves must be routinely disinfected with sterile 70% IPA during the compounding process and must be inspected for holes or punctures.
7. On exiting the compounding area, a person’s gown may be retained for reuse if not visibly soiled, but shoe covers, hair covers, facemasks, and gloves must be replaced with new ones prior to reentry.
8. The preceding activities are not required for preparation of immediate-use CSPs when using a CAI if written manufacturer documentation is available to show that such personnel procedures are not required to ensure an aseptic environment.
H. Competency evaluation of garbing and aseptic work practices
1. Glove fingertip sampling must be used as a tool to evaluate personnel involved in compounding of all CSP risk levels, because touch contamination is the most common breach in aseptic technique.
2. All personnel must be visually observed to confirm competency in hand hygiene, garbing procedures, and routine disinfection of gloved hands.
3. All personnel must pass three gloved fingertip-sampling procedures, with no growth on sterile contact agar plates, prior to being allowed to compound CSPs.
4. After completing the hand hygiene, gowning, and gloving procedures, a gloved fingertip and thumb sample from each hand of the compounder must be collected by having him/her lightly press into the appropriate agar plates.
5.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


