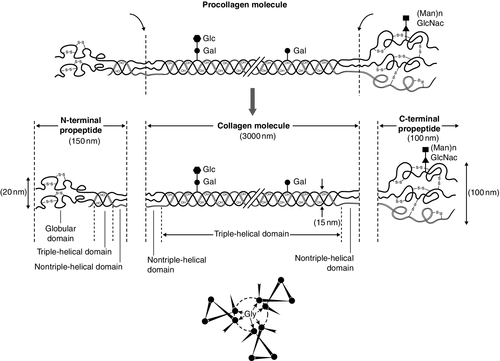
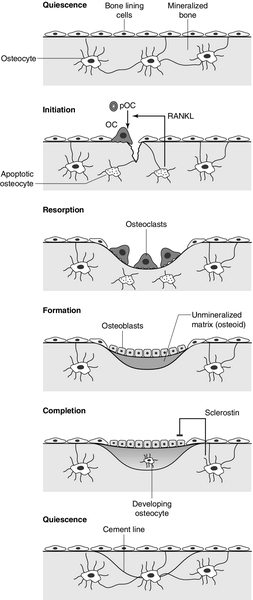
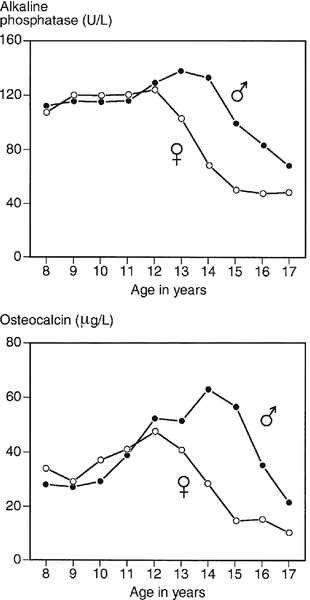
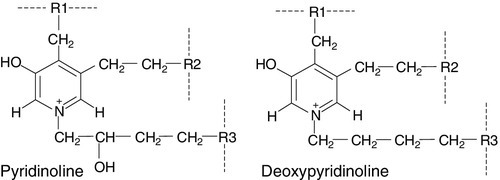
CHAPTER 31 Timothy Cundy; Ian R. Reid; Andrew Grey CHAPTER OUTLINE Biochemical markers of bone turnover Defective osteoblast function and osteomalacia CHRONIC KIDNEY DISEASE – MINERAL AND BONE DISORDER Bone disease after renal transplantation BONE DISEASE IN PRIMARY HYPERPARATHYROIDISM Clinical, biochemical and histological features BONE TURNOVER AND BONE DISEASE IN CHILDREN The principal role of the skeleton is a structural one, maintaining body shape, providing protection for internal organs and, together with the neuromuscular system, making locomotion possible. It also has an important secondary role in mineral homoeostasis, functioning as a reservoir for calcium ions in particular. Metabolic bone diseases can affect both these functions. The anatomist classifies bones as being either flat (e.g. skull, scapula, mandible, ilium) or long (e.g. the limb bones). Flat bones result from intramembranous ossification; long bones predominantly from endochondral ossification. A long bone consists of a shaft (diaphysis) broadening at either end into an epiphysis. The transitional zone between the diaphysis and the epiphysis is termed the metaphysis. On sectioning a long bone, two patterns of organization of bone tissue are found. The elements of bone can be packed together without intervening marrow spaces to form cortical or compact bone, or they can form an interlacing meshwork of trabeculae referred to as cancellous or trabecular bone. The diaphysis of the long bone consists mainly of cortical bone, whereas the metaphysis and epiphysis have a greater quantity of trabecular bone, enclosed within a thin cortical envelope. Some 80% of the weight of an adult human skeleton consists of cortical bone. However, the surface-to-volume ratio of trabecular bone is very much higher than that of cortical bone and it is metabolically much more active. At a microscopic level, bone consists of matrix (~35% by volume), mineral (~60%) and cells (< 5%). The matrix is predominantly type I collagen fibres, usually organized in layers within which the fibres are parallel to one another. In adult bone, the fibre orientation varies from one layer to the next and this is referred to as lamellar bone. If deposited along a flat surface, the lamellae will be parallel to that surface, but in cortical bone they are concentrically oriented around a central blood vessel to form the Haversian canal system. When bone formation is rapid (e.g. during growth or fracture healing) collagen fibres may be laid down with more random orientation, producing woven bone. The mineral phase of bone is hydroxyapatite (Ca10(PO4)6 (OH)2). This forms spindle-shaped crystals, which are found in association with the collagen and ground substance. Their orientation is usually parallel to that of the collagen fibres. There are two general cell types in bone: osteoblasts and osteoclasts. Both are found on the bone surface at sites of active remodelling. Osteoblasts are also thought to be the precursor of two cells types that are more widespread: bone lining cells, which are found over inactive bone surfaces, and osteocytes, which are found in lacunae scattered throughout bone. They are thought to be osteoblasts that have been engulfed by the bone that they have formed. They have long cell processes, which are in contact with similar processes arising from other osteocytes or with those of the bone lining cells. The cell processes lie in an interconnecting network of canaliculi that extends throughout bone tissue. The bone lining cells and osteocytes thus delineate an extracellular fluid (ECF) space that is in contact with the bone surface. This space has a volume of 1–1.5 L and a surface area of several thousand square metres. This is the site of mineral exchange between ECF and bone. The size and nature of this exchange is unknown but the fluid within this space, the bone ECF, has an ionized calcium concentration of only 0.5 mmol/L – less than half of that elsewhere in the ECF. Almost 90% of the protein in bone matrix is type I collagen, which is synthesized by osteoblasts. It is a large molecule (MW > 300 000 Da) with a trimeric helical structure. Type I collagen is initially synthesized in the rough endoplasmic reticulum (RER) as a precursor molecule (type I procollagen) that combines two proα1(I) and one proα2(I) peptide chains (coded by COL1A1 and COL1A2, respectively) in a triple helix. Proα1(I) and proα2(I) have similar structures with a core triple helical domain of 1014 amino acids composed of uninterrupted Gly-X-Y tripeptide repeats, where Gly is glycine and X and Y are often proline or lysine, flanked by propeptides at both N- and C-terminal ends. During and after translation, the three chains undergo extensive modification. Prolyl-4-hydroxylase converts virtually all Y-position proline residues to 4-hydroxyproline, an alteration that is essential for thermal stability of the assembled trimer. In the absence of this modification the trimer melts (that is the individual chains unfold from the stable triple helix, at about 27°C, whereas with full hydroxylation, the melting temperature is about 42°C). Some Y-position lysine residues within the triple helical domain are hydroxylated by the enzyme lysyl hydroxylase-1, and glucose and galactose groups added by glycosyltransferases (Fig. 31.1). Hydroxylation of these triple helical residues is part of the pathway to form stable complex intermolecular crosslinks that provide the tensile strength in tissues. Most of these modifications are completed during translation and occur on the individual chains. If there is a delay in triple helix folding, the process can continue and the physical properties of the chains and molecules are altered and contribute to an osteogenesis imperfecta phenotype. FIGURE 31.1 Type I collagen synthesis. Upper panel: schematic representation of the structure of the procollagen molecule, showing cleavage of the amino and carboxy terminal peptides. Glc, glucose; Gal, galactose; Man, mannose; GlcNac, N-acetylglucosamine. Lower panel: view along the axis of the triple helical structure of the collagen backbone, with every third amino acid a glycine residue forming the inner circle. The three chains that form a [proα1(I)2 proα2(I)] trimer interact through regions in the carboxyl-terminal propeptide of each chain. The full length chain must be maintained in an unfolded state while the carboxyl-terminal propeptides fold, associate, and then begin the process of triple helix formation. Propagation of the collagen triple helix requires a number of enzymes and molecular chaperones to ensure correct folding and trimerization. These include peptidyl disulfide isomerase (PDI), which also forms part of the prolyl 4-hydroxylase complex, and is likely to involve prolyl peptidyl cis-trans isomerase B (also known as cyclophilin B). This protein can act on its own, to assist in the folding around prolyl residues, such as those in the carboxyl-terminal propeptide adjacent to cysteine residues, and as part of a complex that includes two additional proteins, cartilage-related protein and prolyl 3-hydroxylase, to modify certain triple helical prolines. The function of this last process is not yet entirely clear but when the complex is missing, the propagation of the triple helix is altered and modification of the chains increased. Disulphide bonds between the carboxyl-terminal region of the chains act to secure the three chains in a trimer, a process requiring protein-disulphide isomerase. Lysine residues outside the major triple helical domain of type I collagen, needed for the formation of mature intermolecular crosslinks, are hydroxylated by lysyl hydroxylase-2. These complex modifications, which are necessary for correct folding, assuring thermal stability of the triple helix and crosslink formation between collagen molecules once they are secreted into the matrix, need to take place in an orderly and timely sequence, and various chaperone proteins, including HSP47 and FKBP65, help regulate this process. Procollagen trimers are then transported via the Golgi network and packaged into membrane-bound organelles where lateral aggregation, the initial phase of fibril formation, occurs. As secretion occurs, the procollagen molecules are further processed into mature type I collagen molecules by proteolytic cleavage of the N- and C-terminal propeptides (by the enzymes ADAMTS-2 (a disintegrin and metalloproteinase with thrombospondin motifs 2) and BMP1 (bone morphogenetic protein 1), respectively). Finally, the trimers are assembled into collagen fibrils and fibres and anchored in those positions by intermolecular lysine-derived crosslinks in a process that is begun by modification of specific residues by lysyl oxidase. These cross-links are initially reducible, but as tissue maturation proceeds, they are converted into non-reducible compounds including hydroxylysylpyridinoline (derived from three hydroxylysine residues) and the less abundant lysylpyridinoline (derived from two hydroxylysine residues and one lysine residue). The latter (also known as deoxypyridinoline) is present in the body almost exclusively in bone tissue and dentine, where it accounts for 21% of the total mature cross-links. The non-collagenous proteins of bone (proteoglycans, glycosylated proteins, RGD (arg-gly-asp)-containing glycoproteins and γ-carboxylated proteins) account for only 10–15% of bone protein mass but, because of their very much smaller size, are as common as collagen in bone on a molar basis. Most are synthesized by osteoblasts, but a number are produced elsewhere in the body and arrive in bone via the circulation. The latter proteins, which include albumin and platelet-derived growth factor, are usually negatively charged and become bound to hydroxyapatite. The most abundant of the bone-derived non-collagenous proteins are osteocalcin, also known as bone Gla-protein, and osteonectin, both of which have a high affinity for bone mineral. is synthesized by osteoblasts as a 75-amino acid precursor known as pro-osteocalcin. The amino-terminal propeptide includes the substrate recognition site for the vitamin K-dependent γ-carboxylase enzyme which γ-carboxylates three glutamic acid residues (17, 21 and 24) in the central region of the osteocalcin molecule, following which the amino-terminal propeptide is cleaved, leaving the 49-residue osteocalcin molecule. The γ-carboxyglutamic acid residues are strongly calcium binding and allow the molecule to bind to hydroxyapatite. The process of γ-carboxylation seems to be impaired in the elderly. The physiological role of osteocalcin is uncertain, but it probably contributes to the regulation of bone remodelling, since deletion of the osteocalcin gene in mice leads to increased bone density. Its synthesis is regulated by 1,25-dihydroxyvitamin D (1,25(OH)2D, calcitriol) and cortisol, and it is incorporated into bone matrix, being especially abundant in cortical bone. Recently, its uncarboxylated form has been implicated in the regulation of intermediary metabolism, including fat mass, glucose tolerance and insulin secretion. The relevance to human physiology of these findings in genetically modified mice remains to be determined. Several proteoglycans, macromolecules that contain acidic polysaccharide side chains, are present in bone. In the early stages of bone formation, versican, a dermatan sulphate proteoglycan, and the glycosaminoglycan, hyaluronan, are present and may act to delineate areas that are destined to become bone. Subsequently, two chondroitin sulfate proteoglycans, decorin and biglycan, are expressed and may influence cellular proliferation and differentiation in the newly forming bone. Fibromodulin, osteoglycin and osteoadherin are other proteoglycans that are expressed in bone matrix, but their functions are less well characterized. The major glycoproteins present in bone are alkaline phosphatase (ALP), which is highly expressed on the osteoblast cell surface, and osteonectin, a phosphorylated glycoprotein that is also present in several other tissues that undergo rapid remodelling. In bone, osteonectin may regulate both cellular activity and matrix mineralization. At least eight non-collagenous glycoproteins expressed in bone contain the arginyl-glycyl-aspartate (RGD) tripeptide as the consensus cell-attachment sequence (Table 31.1). Some are specific to bone (e.g. bone sialoprotein), but most are more ubiquitously expressed (e.g. fibronectin, osteopontin, thrombospondins, vitronectin). Although the role(s) of each individual protein in skeletal physiology is(are) uncertain, it seems very likely that they collectively serve to promote attachment of bone cells to bone matrix (and perhaps thereby regulate their function). The components of the matrix–cell interactions are clearly dependent upon the maturational stage of the remodelled bone. Three matrix non-collagenous proteins, matrix Gla-protein, protein S and osteocalcin (bone Gla-protein), undergo post-translational γ-carboxylation, a process that enhances calcium binding. These proteins may function physiologically as inhibitors of mineral deposition. TABLE 31.1 Major glycoproteins containing the arginyl-glycyl-aspartate (RGD) cell-attachment motif in skeletal tissue Osteoblasts are the cells responsible for the synthesis and maintenance of bone matrix. They are derived from pluripotent mesenchymal progenitor cells that also give rise to adipocytes, chondrocytes and myocytes. Expression of the transcription factors cbfa1 and osterix is critical for commitment to the osteoblast lineage, while expression of peroxisome proliferation activating receptor-γ (PPAR-γ) favours adipocytic over osteoblastic differentiation. A key molecular event in osteoblast proliferation, and perhaps survival, is signalling through the wnt-low density lipoprotein-related receptor 5 (LRP5)-β-catenin pathway. In this system, secreted wnt proteins bind to and activate LRP5 on the cell surface, which in turn activates a series of intracellular events that result in translocation of cytoplasmic β-catenin to the nucleus, where it activates transcription of genes critical for osteoblast growth and function. The pathway is further regulated by secreted proteins that inhibit wnt-induced activation of LRP5, such as sclerostin, Dickkopf (DKK) and frizzled. Thus, humans carrying mutations of components of this signalling pathway that decrease nuclear translocation of β-catenin have decreased bone mass and fractures as a result of impaired bone formation (e.g. WNT1 and most LRP5 mutations), while constitutive activation of the pathway (e.g. LRP5 mutations affecting one particular extracellular domain or SOST mutations) increases bone mass. Other LRP family members are also important in bone physiology. Mutations affecting one particular region of the extracellular domain of LRP4 result in impairment of the ability of sclerostin to inhibit bone formation, and thereby cause increased bone mass (sclerosteosis type 2). Mature osteoblasts are metabolically very active and intimately involved in bone formation. They synthesize many of the matrix proteins referred to above. They have very high levels of alkaline phosphatase (ALP) activity associated with their cell membranes. This enzyme is necessary for normal bone mineralization to take place. Osteoblasts express receptors for parathyroid hormone (PTH) and 1,25(OH)2D, as well as a number of cytokines, growth factors and sex hormones. Osteoblasts also play a key role in regulating osteoclastic bone resorption, ensuring that the components of bone remodelling, formation and resorption are coupled. A critical mechanism by which osteoblasts regulate bone resorption is via the RANK–RANKL–OPG system (see below). Osteocytes and bone lining cells are terminally differentiated osteoblasts derived from mature osteoblasts that are no longer involved in active bone formation, and have become entrapped within the canaliculi of the bone matrix that they have produced. This process of osteocytogenesis is probably an active one, requiring cleavage of matrix proteins: mutations of the metalloproteinase MT1-MMP lead to osteocytes with fewer and shorter dendritic processes. As osteocytogenesis occurs, the osteoblast changes from a cuboidal cell to a typical osteocytic morphology, characterized by a stellate appearance and numerous cytoplasmic projections that permit the cell to communicate with cells on the bone surface and other osteocytes living within adjacent bone. Concurrently, the genetic signature of the cell changes from a typically osteoblastic one to that which reflects a mature osteocyte, in particular the expression of DMP1, SOST and FGF23. Osteocytes play a key role in the regulation of bone remodelling. At sites of skeletal microdamage, osteocytes undergo apoptosis and release apoptotic bodies expressing RANKL (see below), promoting the recruitment of osteoclasts to the area to initiate remodelling to allow repair of the damaged bone. Osteocytes also produce and release sclerostin, an endogenous inhibitor of osteoblast differentiation and function that acts to inhibit wnt-induced activation of LRP5. They also act as mechanosensory cells in bone, perhaps in tandem with osteoblasts lining the bone surface, with which they communicate. Finally, osteocytes are critically important for bone mineralization, by regulating phosphate metabolism at a whole organism level, principally by production of the phosphaturic hormone fibroblast growth factor 23 (FGF23), a process that involves the osteocyte products phosphate-regulating endopeptidase (PHEX) and dentine matrix phosphoprotein 1 (DMP1). Osteoclasts are the cells that resorb bone. They are derived from cells of the monocyte/macrophage series. They are multinucleated and are usually found singly or in small numbers within resorption lacunae of their own making. They have large numbers of mitochondria and lysosomes. Activated osteoclasts possess a ‘ruffled’ border, adjacent to the bone being resorbed. The ruffled border is delimited by a ‘sealing zone’ at which the osteoclast is attached to the underlying bone. Bone resorption occurs as a result of the secretion of protons and proteolytic enzymes into the space between the ruffled border and the bone surface. Among these enzymes are tartrate-resistant acid phosphatase (a useful histological marker of osteoclasts) and cathepsin K. Osteoclast development and function are regulated by osteoblast and osteocyte-derived cytokines, in particular osteoprotegerin (OPG) and receptor activator of nuclear factor-κB ligand (RANKL), which are members of the tumour necrosis factor (TNF) receptor superfamily. RANKL expressed on the osteoblast and osteocyte surface binds to, and activates its receptor – receptor activator of nuclear factor-κB (RANK) – which is expressed on the surface of osteoclast precursors. RANKL is also shed from the cell surface, and this soluble form of the protein may confer osteoclastogenic activity distant from the cell of origin, in particular the osteocyte. Interaction between RANKL and RANK promotes osteoclastogenesis. Osteoprotegerin is a secreted osteoblast product that acts as a decoy receptor for RANKL, and thereby functions as an endogenous inhibitor of osteoclastogenesis. Thus, the relative levels of expression by the osteoblast of RANKL and OPG determine the rate at which osteoclastogenesis occurs. Many of the systemic factors that alter bone resorption, such as PTH and 1,25(OH)2D (which increase osteoclast function) and sex hormones (which decrease it), do so by changing osteoblastic production of RANKL and/or OPG. An exception is calcitonin, whose receptor is expressed on osteoclasts, and which therefore inhibits bone resorption directly. A number of other critical cellular and molecular events are now recognized to underpin osteoclastic bone resorption. The intracellular tyrosine kinase c-src regulates osteoclast motility and aspects of cytoskeletal function that are critical to formation of a functional resorption space; the αvβ3 integrin (vitronectin receptor) plays a key role in osteoclast attachment to bone; the cysteine protease cathepsin K and the type 7 transmembrane chloride channel (ClCN7) are each required for proteolytic degradation of bone matrix. In mature bone, there is a continuous process of removal and replacement of pockets of old bone. This is termed bone remodelling and is shown in schematic form in Figure 31.2. The process occurs at discrete sites on the bone surface and, in the mature skeleton, is probably an adaptive response to skeletal microdamage. Bone remodelling is achieved by teams of osteoclasts and osteoblasts acting within the anatomical structure known as the basic multicellular unit (BMU). Osteocytes play a critical role in initiating bone remodelling, and probably also in regulating the coordination of formation and resorption within the BMU. Thus, apoptotic osteocytes in an area of skeletal microdamage produce RANKL that directs osteoclastogenesis and initiation of bone remodelling to the damaged bone. At that site, the osteoclasts excavate a resorption pit, the depth of which is determined by the number and activity of the osteoclasts present. The motile osteoclasts are then replaced by osteoblasts, which proceed to fill the resorption pit with bone matrix. Mineralization of this new bone then occurs, lagging behind matrix synthesis by a period of about three weeks. Once the resorption pit has been filled, the osteoblasts either undergo apoptosis, return to their quiescent state as bone lining cells or survive as osteocytes within the bone lacunae. FIGURE 31.2 The bone remodelling cycle. Bone remodelling is initiated in response to skeletal microdamage, sensed by adjacent osteocytes which undergo apoptosis and produce the osteoclastogenic cytokine, receptor activator of nuclear factor-κB ligand (RANKL). Osteoclastic resorption of the damaged bone ensues, after which mature osteoblasts progressively fill the resorption cavity with osteoid, and coordinate its subsequent mineralization. Some of the osteoblasts develop into osteocytes within the lacunae of the newly formed bone. Sclerostin is produced by osteocytes adjacent to the remodelling space: it signals inhibition of bone formation and a return to a quiescent state. In skeletal homeostasis, the amount of new bone formed equals that resorbed during each remodelling cycle. OC, osteoclast; pOC, pre-osteoclast. (Courtesy of Dr Andrew Grey.) An increase in bone turnover is characterized by a faster bone remodelling cycle initiation rate, rather than an increase in the duration of the cycle. The histological consequence of such an increase is that a greater proportion of bone surface is involved in remodelling at any one time. The time required to complete a full cycle of resorption and formation (σ) is in the order of 6–12 months. Inhibiting bone resorption, for example with pharmaceutical agents, rapidly reduces the initiation rate of new remodelling cycles, but those cycles already initiated go to completion. Thus there is a period (‘a transient’) of duration σ, when the bone formation rate exceeds the resorption rate. This ends when all those cycles initiated before the inhibition of bone resorption took place have completed their formation period. The bone formation rate at this stage will have fallen to match the new, lower, resorption rate. Bone resorption and formation are thus very closely linked in normal bone and also in many metabolic bone disorders. As outlined earlier, the osteoclasts and osteoblasts clearly ‘communicate’ with each other. Osteoblastic regulation of osteoclast function is mediated in large part by the RANK–RANKL–OPG system, but the nature of the osteoclast-derived signal(s) that recruits and activates osteoblasts is not yet fully characterized. Osteocyte-derived sclerostin, an inhibitor of osteoblast differentiation, probably contributes to the regulation of the bone formation component of the remodelling cycle. Matrix-associated growth factors that are released from resorbed bone, such as transforming growth factor β (TGFβ), bone morphogenetic proteins (BMPs) and platelet-derived growth factor, may also constitute part of this signal. Finally, genetic evidence suggests that communication from osteoclasts to osteoblasts occurs via members of the ephrin (Eph) family of signalling proteins, specifically osteoclast-expressed ligand ephrin-B2 signalling through the osteoblast-expressed receptor EphB4 to activate osteoblast differentiation and bone formation. During growth, the amount of bone deposited exceeds that which is removed and there are also changes in the size and configuration of bones (modelling). In senescence, because of factors including sex hormone deficiency, reduced physical activity and various endocrine and inflammatory diseases, the balance of formation and resorption is reversed and bone is lost. Age-related bone loss affects both cortical and trabecular bone, but early after the menopause, bone loss is particularly rapid from the axial skeleton, which has a greater proportion of trabecular bone. A large number of factors have been found to influence bone remodelling (discussed in Chapter 6). Bone turnover is a term referring to both osteoblastic bone formation and osteoclastic bone resorption. These processes can be assessed by bone histomorphometry, skeletal scintigraphy and by analysing the kinetics of intravenously administered isotopes of calcium. There are numerous biochemical markers of bone turnover, representing either products of bone cells or degradation products from the breakdown of bone collagen (Box 31.1). The number of potential markers available has increased considerably in recent years. In a small number of rare conditions (e.g. hypophosphatasia, osteopetrosis, Bruck syndrome and mild osteogenesis imperfecta), a specific pattern of bone turnover markers can be helpful in diagnosis. However, the main use of markers is to establish whether a high bone turnover state exists. High bone turnover, with its various adverse consequences, is a feature of many of the more common metabolic bone disorders, and a key target of currently available therapies. Alkaline phosphatase is widespread in human tissues. Enzymes with this activity are coded for by genes at three separate loci, their respective products being referred to as intestinal, placental and bone/liver/kidney (or tissue non-specific) ALP. The latter gene is located on chromosome 1p36. The gene transcript has a number of glycosylation sites, and the bone, liver and kidney isoenzymes differ in their carbohydrate side chains. The major limitation of total ALP as an index of bone formation is its multiple tissues of origin and hence its lack of sensitivity at low values. In health, plasma ALP activity is principally derived from the liver and osteoblasts in approximately equal proportions. If the activity of other hepatic canalicular cell enzymes is normal, then elevated total ALP activity usually reflects osteoblastic activity (or, more precisely, the number of active osteoblasts). If uncertainty persists in determining the origin of increased plasma ALP, then isoenzyme studies may be undertaken. The bone and liver isoenzymes have differing stabilities to heating (the bone fraction being more sensitive to heat inactivation) and different mobilities on electrophoresis. Immunoassays for bone-specific ALP use isoform-specific monoclonal antibodies; even so, there is up to 20% cross-reactivity with the liver isoform in these assays. Thus very high plasma activities of liver ALP, as occur in hepatobiliary disease, can still interfere with the measurement of bone-specific ALP. The activity of the bone isoenzyme has a diurnal variation in plasma, with peak values occurring around midnight and being some 25% higher than early morning values. Although the half-life in plasma of the bone isoenzyme is very short (~2 days), in metabolic bone disorders changes in ALP activity reflect slower processes – the acceleration or deceleration of the remodelling cycle – so the rate of change of plasma ALP is also much slower. The marked elevation in plasma ALP activity seen in childhood and adolescence is due to an increase in the bone fraction. The plasma ALP activity closely parallels the growth velocity curve (Fig. 31.3). A significant (40–50%) rise in plasma ALP activity occurs at the menopause because of accelerated bone turnover. Plasma activity of the enzyme can also be increased after major fractures and reflects the increased cellular activity associated with healing. After hip fracture, for example, ALP activity reaches a peak at around four weeks, with values approximately double those on admission to hospital. Total plasma ALP activity is increased during pregnancy because of the appearance of the placental isoenzyme. FIGURE 31.3 Bone turnover markers during growth. Mean values of plasma alkaline phosphatase (ALP) and osteocalcin in males ([closed circle]) and females ([open circle]) through childhood and adolescence. Peak values are reached at an average age of 12 years in girls and 14 in boys, coinciding with peak growth velocity. As growth velocity falls, both these indices decline toward adult values. (Data from Round JM 1973 Plasma calcium, magnesium, phosphorus and alkaline phosphatase concentrations in normal British school children. British Medical Journal 3: 173–140, and Johansen JS, Riis BJ, Podenphant J, Skakkeboek NE, Christiansen C 1987 Plasma bone Gla protein: a specific marker of bone formation. In: Christiansen C, Johansen JS, Riis BJ (eds). Osteoporosis, vol 2. Copenhagen: Osteopress ApS: 677–681.) The entity of familial benign hyperphosphatasia is characterized by asymptomatic high ALP activities (predominantly of intestinal isoform) and dominant inheritance, but with no clinical evidence of bone disease. Benign transient hyperphosphatasaemia is a condition characterized by high concentrations of both bone and liver isoenzymes, first identified in children but now recognized in adults (see Chapter 13). There are a number of causes of low ALP activities (hyophosphatasaemia), including vitamin C deficiency, hypothyroidism, starvation, zinc or magnesium deficiency, Cushing syndrome, Wilson disease and the genetic disorder, hypophosphatasia (discussed below). Osteocalcin is produced by osteoblasts, and is generally regarded as a marker of bone formation, but it seems to be involved in the process of mineralization rather than matrix production. Plasma concentrations of intact osteocalcin are markers of bone formation, and circulating concentrations generally correlate well with histological measures of bone formation rate. Around 25% of circulating osteocalcin consists of intact osteocalcin, the remaining immunoreactivity comprising N-terminal, mid-region, mid-region-C-terminal and C-terminal fragments. When bone is resorbed, osteocalcin fragments are released, and the plasma concentrations of these fragments may thus reflect bone resorption. These fragments are cleared predominantly by the kidneys, so there is considerable immunological heterogeneity in plasma osteocalcin in patients with renal impairment. These factors may limit the usefulness of osteocalcin as a marker of bone formation. Osteocalcin has a particular advantage in having greater sensitivity than ALP in the detection of low rates of bone formation. As with plasma ALP activity, plasma osteocalcin and growth rate are closely correlated (see Fig. 31.3), and there is a two-fold rise in plasma osteocalcin concentration at the menopause. Plasma osteocalcin concentrations fall markedly during pregnancy. Cord blood concentrations are two to three times higher than adult values. The half-life of intact osteocalcin in the circulation is about 5 min. Plasma osteocalcin concentrations exhibit a diurnal variation with the peak in the early morning being about 15% higher than the nadir value, which occurs in the afternoon. Concentrations may also be depressed (by about 10%) by alcohol intake and are markedly suppressed (by about 50%) by glucocorticoids. Plasma concentrations rise when osteoblasts are stimulated by the administration of 1,25(OH)2D. Antibodies have been raised to both the soluble C-terminal and N-terminal propeptides of procollagen, which appear in the circulation after cleavage from the newly formed collagen molecule. Their concentrations in the plasma thus reflect osteoblastic collagen synthesis. As they are not filtered by the kidney, variations in renal function should not affect their plasma concentrations. Degradation of the proteins takes place in hepatic endothelial cells (through the mannose 6-phosphate receptor in the case of procollagen 1 carboxy-terminal extension peptide (P1CP), and by scavenger receptors in the case of procollagen 1 amino-terminal extension peptide (P1NP)). Procollagen 1 carboxy-terminal extension peptide is usually measured by enzyme linked immunosorbent assay (ELISA). It has a diurnal variation, with peak concentrations occurring at 04.00–08.00 h, and the nadir in the afternoon. There may be some contribution to circulating P1CP from non-osseous sources. Procollagen 1 amino-terminal extension peptide has been more widely studied. It appears to be a more dynamic marker than P1CP or other formation markers. The peptide is stable, and the available assays are precise and responsive to common interventions in metabolic bone diseases. It is now regarded as the marker of choice to assess osteoblast activity. Of the various assays available for P1NP, some measure the intact propeptide (intact P1NP) while others (total P1NP) also detect a smaller antigen in serum. In many clinical situations, these assays give similar information, but renal insufficiency increases the concentrations of the smaller antigen, influencing both the apparent concentration of P1NP and assay calibration. Hydroxyproline is a major component of fibrillar collagen of all types, comprising ~14% of the total amino and imino acid content. It is produced by the post-translational modification of proline by the enzyme 4-prolyl hydroxylase. In plasma, hydroxyproline exists in protein-bound, peptide-bound and free forms. Most hydroxyproline is oxidized in the liver but a small proportion (~10%) is excreted in the urine. Approximately 90% of collagen-derived hydroxyproline excretion is in the form of peptides resulting from collagen degradation, with the remainder in the form of fragments of the N- and C-terminal procollagen peptides cleaved during collagen synthesis. When bone resorption is increased, urinary hydroxyproline excretion rises, because of the increased rate of catabolism of type 1 bone collagen. When bone turnover is low, urinary hydroxyproline is not a reliable index of bone resorption, since the other sources (for example C1q complement component) can contribute up to 50% of the total urinary hydroxyproline. Like all urinary collagen breakdown products, hydroxyproline excretion can be measured in a 24 h urine collection or, more conveniently, in fasting, second voided morning samples, expressed as a ratio to the creatinine concentration. Dietary sources of hydroxyproline (e.g. gelatin) need to be eliminated immediately before and during the collection. The major limitations of hydroxyproline estimation as an index of bone resorption are interference from sources other than type 1 collagen, its lack of sensitivity at low concentrations, its day-to-day variability and its partial origin from bone formation. Hydroxylysine – another amino acid unique to collagen-like peptides – is produced by the post-translational modification of lysine residues. These amino acids can then undergo further modification by glycosylation – giving rise to galactolysyl-hydroxylysine and glucosylgalactolysyl-hydroxylysine. The former predominates in the type 1 collagen of bone. Galactolysyl-hydroxylysine is not significantly metabolized before excretion in the urine (10% in free form, 90% peptide bound), and the urinary concentrations are not affected by diet, so it is a good marker of bone resorption. It can be measured by HPLC, but the assay is technically demanding. The amino acids hydroxylysylpyridinoline and lysylpyridinoline (also known as pyridinoline and deoxypyridinoline, respectively) are created by the covalent cross-linking that takes place when mature collagen molecules stabilize after forming the triple helical structure (Fig. 31.4). Since the cross-links are formed only in extracellular collagen fibrils, they appear in the circulation and the urine only as degradation products of mature matrix and do not reflect new collagen synthesis. In contrast to hydroxyproline, neither pyridinoline nor deoxypyridinoline occur in the collagen of normal skin. Pyridinoline is present in other connective tissues, notably cartilage and tendon, whereas the less abundant deoxypyridinoline is more specific to bone and dentine. FIGURE 31.4 Molecular structure of pyridinoline cross-links. R1 and R2 are telopeptide sequences and R3 a helical fragment sequence; for free crosslinks, R1, R2 and R3 are: − CH(COOH)NH2. In urine, both pyridinoline and deoxypyridinoline are present as both free amino acid derivatives (~40%) and as oligopeptide-bound fractions (~60%). The free forms can be measured directly, but the conjugated form has to be hydrolysed before assay. After acid hydrolysis, total urinary pyridinoline and deoxypyridinoline can be measured by HPLC, but ELISA immunoassays for the free amino acids or peptides containing the cross-links are now available. The pyridinoline:deoxypyridinoline molar ratio in urine is similar to that found in bone (7:2), and the urinary excretion rate of the cross-links agrees well with radioisotopic and histomorphometric measures of bone resorption. The cross-links do not undergo further metabolism once released, and their rate of excretion is not influenced by dietary intake or moderate renal impairment. Random urine measurements (expressed relative to creatinine concentration) correlate closely with 24 h estimates. The elderly tend to have relatively greater excretion of the larger peptide-bound forms, and a correspondingly smaller proportion of the free pyridinoline and low molecular weight peptide forms. Urinary pyridinoline and deoxypyridinoline concentrations appear to be good markers of bone resorption, and more sensitive than hydroxyproline in distinguishing small increases in bone resorption from normal. For example, when normal premenopausal and postmenopausal women are compared, hydroxyproline excretion in the latter group is about 50% above that of premenopausal women, whereas for urinary pyridinoline and deoxypyridinoline, excretion is increased by 100%. Collagen cross-links form mainly between the short non-helical peptides at both ends of the collagen molecule (the N- and C-terminal telopeptides), which link via pyridinium or pyrrole compounds to the helical portion of an adjacent collagen molecule (Fig. 31.5). During collagen breakdown, N- and C-terminal telopeptide fragments, still attached by the pyridinium cross-link to the helical fragments of the nearby molecule, are released into the circulation and are cleared by the kidneys. Within the C-telopeptides of type I collagen is an aspartyl-glycine motif that can undergo β-isomerization to isoaspartyl-glycine. This process is believed to be linked to protein ageing, with younger bone having a greater proportion of the non-isomerized α-C-telopeptides, and older bone having a greater proportion in the β-isomerized C-telopeptide form. Various immunoassays have been developed to detect epitopes specific to particular fragments. FIGURE 31.5 Intermolecular cross-linking in type I collagen and the location of the peptide sequences of the N-terminal (NTX) and C-terminal (CTX) immunoassays. The urinary N-telopeptide (NTX) assay is an ELISA based on a monoclonal antibody that recognizes a conformational motif in the α2-chain that is linked to a pyridinium cross-link. Although it does not recognize the pyridinoline or deoxypyridinoline cross-link itself, it is specific for the cross-linking site of bone collagen. The assay requires no hydrolysis. The measurement is usually performed on a second void, morning urine sample, and the results are expressed as bone collagen equivalent, corrected for urinary creatinine concentration. A serum assay has also been developed, which correlates well with urinary concentrations. Immunoassays for the C-terminal telopeptide cross-linked region of type I collagen (CTX) have also been developed. The monoclonal antibody recognizes an octapeptide sequence of the α1 chain containing the lysine in the C-terminal telopeptide that is involved in intermolecular cross-linking. Immunoassays that are specific for either α-CTX or β-CTX have been developed, and can measure these fragments in both serum and urine. Serum β-CTX is generally regarded as the marker of choice for assessing bone resorption. It should be measured fasting at 08.00–09.00 h, since it shows diurnal variation and is suppressed by food. The acid phosphatases are lysosomal enzymes that hydrolyse phosphomonoesters at low pH, releasing phosphoric acid. They are produced in a number of tissues, including bone, prostate, platelets and erythrocytes. Five isoenzymes are detectable in plasma. The different acid phosphatase isoenzymes differ in tissue of origin, chromosomal origin, molecular size and electrophoretic mobility. Based on the latter, the plasma isoenzymes have been classified as types 1–5. According to their sensitivity to inhibition by L(+)-tartrate, the isoenzymes are further classified as tartrate sensitive (TSAP) and tartrate resistant (TRAP). Prostatic acid phosphatase (type 2b) activity is typically raised in plasma in carcinoma of the prostate. Type 5 acid phosphatase is produced by macrophages (TRAP5a) and osteoclasts (TRAP5b). TRAP5a activity in plasma is elevated in Gaucher disease and hairy cell leukaemia. Tartrate resistant 5b differs from TRAP5a by not having sialic acid residues. It can be measured in serum by a variety of kinetic assays or by immunoassay. Measurement has some value in the diagnosis of osteoclast-rich forms of osteopetrosis. In most forms of this disorder, the number of osteoclasts in bone is actually increased – although they are ineffective at resorbing bone – and plasma activities of TRAP5b are increased. In most other bone disorders, TRAP5b plasma concentrations correlate well with other indices of bone resorption and with histological measurements of osteoclastic activity. Unlike other resorption indices, TRAP5b reflects osteoclast number and/or activity, rather than matrix breakdown. It may find a particular role in situations where these are dissociated, such as in examining the actions of drugs that inhibit osteoclast activity without reducing their number (e.g. cathepsin K inhibitors). Plasma acid phosphatase concentrations are also increased in bisphosphonate-induced osteosclerosis. It is now possible to measure circulating cathepsin K concentrations. Preliminary reports indicate that these are higher in subjects with fractures, and are related to other turnover markers. Assays are also available for circulating osteoprotegerin, soluble RANKL, and sclerostin. These factors all act locally in bone, and the relevance of circulating concentrations to judging their bioactivity in the bone micro-environment remains to be determined. These new markers are used mainly for research purposes at present, and their clinical utility is uncertain. Many of the markers described are now widely used in research, and are finding a place in the management of some metabolic bone diseases. In general, they correlate well with assessments of turnover based on histomorphometry or calcium kinetic studies. The newer assays are more specific to bone and, in some cases, are more sensitive to perturbations, but their usefulness is limited by the day-to-day biological variability (as well as analytical imprecision). Stability is also an important issue for some analytes. In general, plasma markers of bone resorption show less day-to-day variability than urinary markers. Urinary resorption markers are usually measured on fasting second void urine samples to minimize the effect of diet and results expressed as a ratio to creatinine concentration. Thus, artefact can arise if muscle mass is significantly altered from normal. Most markers of bone turnover show significant diurnal variation with highest values in the early morning hours and lowest values during the afternoon or night. The amplitude of this variability is around 15–30%. Therefore, resorption markers are best assessed in the early morning, after overnight fasting. Bone turnover may also vary with the menstrual cycle: formation markers are higher in the luteal phase, whereas resorption markers are higher during the follicular phase. The amplitude of these changes is ~10%. After the menopause, there is an increase in bone turnover – resorption markers increase by 30–50% within 12 months of the menopause, and there are similar, but later, increases in formation markers. A less marked increase in bone turnover markers is also seen in men after the age of 50 years. Compared with adult values, all turnover markers are much higher during childhood and puberty, because the skeleton is growing and modelling, as well as remodelling (see below). It is thus important that age-appropriate reference ranges are used for all these assays. Bone markers increase by ~50% two to four weeks after a fracture, and can take six to nine months to return to normal. Osteoporosis is a reduction in bone density to the point that fractures occur as a result of the minor trauma that is a normal part of everyday life. At the microscopic level, osteoporotic bone is normally mineralized but reduced in volume. There is a generalized thinning of trabecular elements with the total loss of some trabeculae, greatly reducing the strength of bone (Fig. 31.6) and a reduction in cortical width in long bones. This loss of the normal microarchitecture emphasizes the need for prevention of bone loss, since restoration of the lost structure is unlikely to be achievable with pharmacological agents. FIGURE 31.6 Low-power scanning electron micrographs of iliac crest biopsies at the same magnification. (A) Taken from a healthy 47-year-old woman; (B) from an osteoporotic woman of 75 years. In comparison with (A), there has been thinning of the trabeculae with loss of some trabecular elements and discontinuity (arrowed) of some of those which remain. (From Dempster D W et al. 1986 A simple method of correlative light and scanning electron microscopy of human iliac crest bone biopsies. Journal of Bone and Mineral Research 1:21–25, with permission.) Osteoporosis can also be defined using bone mineral density (BMD) criteria. Thus, osteoporosis is defined as a BMD T-score less than − 2.5, a BMD that is more than 2.5 standard deviations below the mean value in the young healthy population. Using this diagnostic criterion, ~50% of women have osteoporosis by the age of 80 years. Osteopenia is defined as a BMD T-score that is 1–2.5 standard deviations below the mean value in the young healthy population. Low bone mineral density is an important predictor of future fractures. Loss of BMD is sometimes pathological, but more frequently occurs as a normal part of ageing. With age, the efficiency with which osteoblasts refill resorption cavities is reduced, and there is also an increase in bone resorption associated with hypogonadism. The important clinical consequence of osteoporosis is fracture. Common osteoporotic fractures include those of the hip, forearm, vertebral body, humerus, ribs and pelvis, while digital, skull, facial and fibular fractures are generally not osteoporotic in origin. The epidemiology of osteoporotic fractures reflects the importance of age and sex in the pathogenesis of the disease. The incidence of vertebral and hip fractures increases exponentially in later life in both men and women, although fracture rates at these sites in men increase about 10 years later (at age 75) than those in women. Box 31.2 sets out a list of risk factors for, and causes of, osteoporosis. Some of these (for example age and family history) are not modifiable. Whether or not an individual will develop osteoporosis depends on two factors: the peak bone mass, typically attained by age ~20 years, and the rate of loss of bone in later life. Probably the strongest influence on peak bone density is genetic – there is a close relationship between an individual’s BMD and that of his parents, and bone density is reduced in the children of patients with osteoporosis. Because of the evidence for a strong heritable component to peak BMD, there has been intense interest in identifying genes that regulate skeletal health. Thus, a large number of genome-wide association studies (GWAS) have been performed in recent years. These analyses have identified more than 20 genes that are associated with alterations in BMD. Several of these genes cluster into four biological pathways: the vitamin D endocrine pathway, the oestrogen endocrine pathway, the wnt-β-catenin signalling pathway and the RANKL–RANK–OPG pathway. However, collectively these genes account for only a few percent of the variability in BMD. So, at present, it seems unlikely that genetic testing will improve the predictive power of algorithms used to identify patients at high fracture risk. Genetic factors may also underlie the significant racial differences in bone density and fracture incidence. Individuals of African or Polynesian ancestry have higher bone mass than Europeans and Asians.
Metabolic bone disease
BONE BIOLOGY
Anatomy of bone
Macroscopic
Microscopic
Bone matrix proteins
Collagen
Non-collagenous proteins
Osteocalcin
Other bone proteins
Protein
Proposed function
Thrombospondins
Cell attachment, bind heparin, collagens, thrombin, fibrinogen, plasminogen
Fibronectin
Binds to cells, collagen, fibrin, heparin, gelatin
Vitronectin
Cell attachment, binds collagen and plasminogen
Fibrillin
Regulation of elastic fibre formation
Osteopontin
Binds to cells, inhibits mineralization, regulates cell proliferation and tissue repair
Bone sialoprotein
Binds cells, may inhibit mineralization
BAG-75
Binds calcium,?cell attachment
Dentin matrix phosphoprotein 1
Osteocyte-derived, regulates osteocyte maturation and FGF23 production
Cellular elements of bone
Osteoblasts
Osteocytes
Osteoclasts
Bone remodelling and its regulation
Biochemical markers of bone turnover
Markers of bone formation
Alkaline phosphatase
Osteocalcin
Procollagen 1 extension peptides
Markers of bone resorption
Hydroxyproline
Glycosylated hydroxylysine
Collagen cross-links
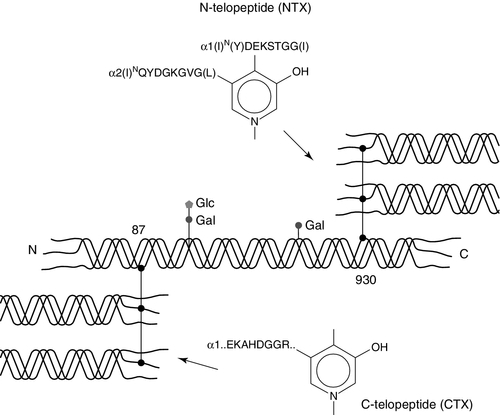
Collagen telopeptides
Tartrate-resistant acid phosphatase
New markers
Variation in bone turnover markers
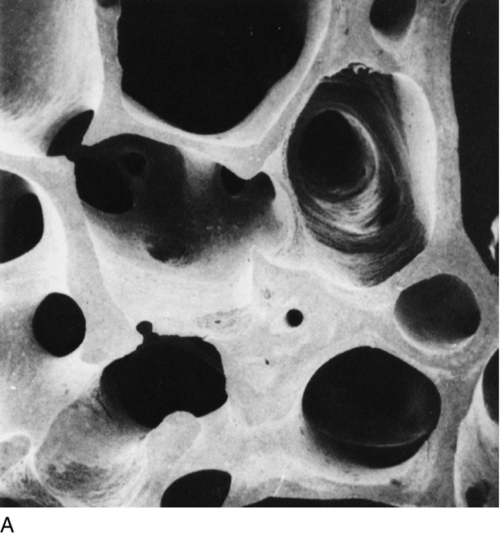
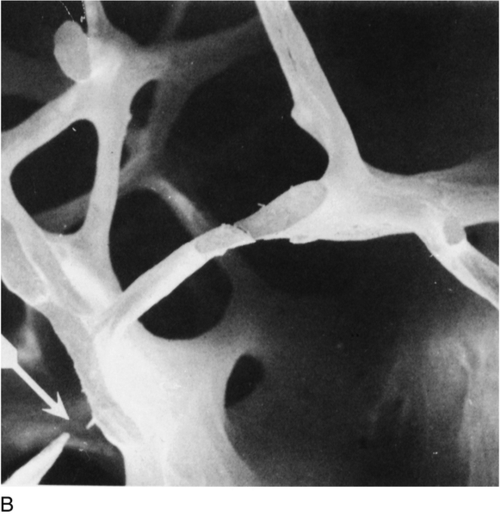
OSTEOPOROSIS
Causes of osteoporosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


