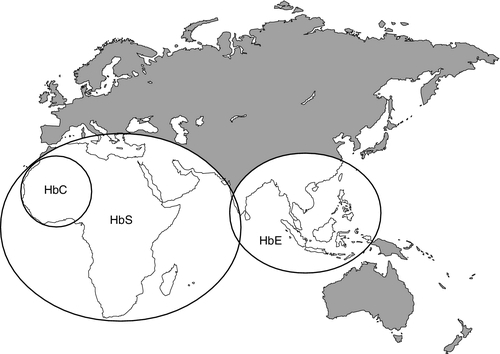
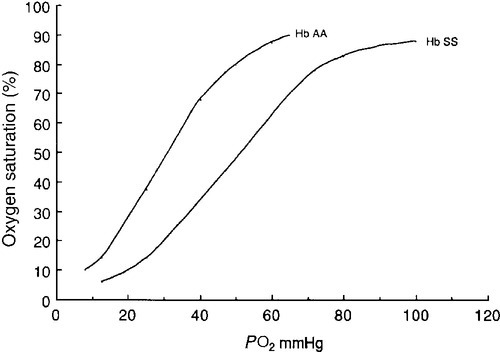
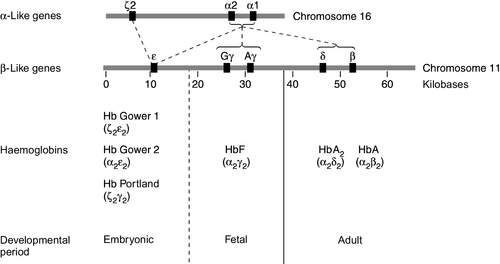
CHAPTER 29 CHAPTER OUTLINE The structure and function of haemoglobin The genetic control of haemoglobin synthesis STRUCTURAL HAEMOGLOBIN VARIANTS Other structural haemoglobin variants LABORATORY DIAGNOSIS OF HAEMOGLOBINOPATHIES Since the discovery of its central role in oxygen transport in the 19th century, haemoglobin has proved to be a source of fascination to scientists and physicians alike, and is arguably the most widely studied protein in man. Its principal disorders, the thalassaemias and sickle cell disease, quantitative and qualitative defects of synthesis, respectively, are among the most common serious inherited diseases in man and have been pivotal in efforts to understand the molecular basis of human diseases. It is estimated that about 7% of the world’s population carry the mutation for at least one type of haemoglobinopathy, and that 400 000 babies are born each year with a severe form. Approximately 230 000 babies are born each year with sickle cell disease in sub-Saharan Africa, and 120 000 are born with severe thalassaemia in southern and South-East Asia. There is good epidemiological and experimental evidence that the extraordinarily high frequency of haemoglobinopathies and their geographical distribution are due to the relative protection that they provide against malaria and its serious complications. This principally involves improved survival and reproductive success for heterozygotes compared with the normal population, which outweighs the reduced reproductive success of homozygotes. Thalassaemia reaches polymorphic frequencies (> 1%) in nearly all populations, except those of northern Europe, northern Asia and the indigenous peoples of Australia, the Americas and the Arctic. The common abnormal haemoglobins have more limited distributions (Fig. 29.1). FIGURE 29.1 Primary geographic distribution of haemoglobins (Hb) S, C and E. Since there is considerable overlap in the world distribution of thalassaemias as well as the common structural haemoglobin variants, co-inheritance of more than one haemoglobin abnormality is common. This generates an extremely diverse range of clinical phenotypes. Haemoglobin is a tetramer comprising two α and two β globin chains (α2β2). One molecule of haem, which contains iron and can bind to oxygen, is attached to each globin chain, lying in a hydrophobic cleft. The amino acid sequence of this haem pocket shows marked homology between different animal species, suggesting a specific and important function. Through its iron atom, each haem group is capable of binding one molecule of oxygen. The reversible binding of oxygen to haemoglobin is allosteric, giving rise to the characteristic shape of the oxygen dissociation curve (Fig. 29.2). On binding oxygen, conformational changes occur in both individual globin chains and the tetrameric structure of haemoglobin. The predominant change is closure of the gap between the two β-chains. The site at which this occurs is of critical functional importance. 2,3-Diphosphoglycerate (2,3-DPG), the principal regulator of oxygen affinity in red cells, binds to this part of the haemoglobin molecule. This serves to separate the two β-chains, favouring the deoxygenated conformation and thereby reducing the oxygen affinity of haemoglobin. The observation that oxygen affinity is often reduced in anaemic patients is explained by the adaptive increase in intracellular 2,3-DPG concentration. This enhances oxygen delivery to the tissues and is one of the important reasons why patients frequently tolerate chronic anaemia with very few symptoms. Conversely, the higher oxygen affinity of fetal blood necessary to maintain adequate maternal–fetal oxygen transport is, in large measure, due to the lower affinity of 2,3-DPG for fetal haemoglobin (HbF). FIGURE 29.2 The oxygen dissociation curves of normal (AA) and sickle (SS) haemoglobin (Hb). The other main physiological influence on haemoglobin oxygen affinity is the Bohr effect. First recognized at the turn of the century through the effect of carbon dioxide on lowering the oxygen affinity of whole blood, it is now known to reflect the sensitivity of oxygen binding by haemoglobin to changes in blood pH. An increase in hydrogen ion concentration stabilizes the deoxygenated conformation of the haemoglobin molecule, and so physiological acidosis leads to a reduction in the oxygen affinity. If acidosis is sustained, this effect may be counterbalanced by the reduction in intracellular 2,3-DPG concentration due to modulation of red cell glycolysis. Several inherited variants of haemoglobin have been described that stabilize the molecule in either the oxy or deoxy conformation. In some cases, the interaction with 2,3-DPG or the Bohr effect is modified. A further factor affecting haemoglobin function is temperature. An increase in temperature reduces oxygen affinity, whereas a fall increases it. As the human is normally isothermic, this is usually of little consequence in terms of physiological adaptation, though during exercise, when oxygen consumption is increased, a raised temperature in the muscles together with an increase in hydrogen ion concentration favours release of oxygen by haemoglobin. Extreme hypothermia has many adverse consequences, including increased haemoglobin oxygen affinity causing reduced oxygen delivery to tissues. Human globins are encoded by the α and β gene clusters located on chromosomes 16 and 11, respectively (see Fig. 29.3). The α-like genes include an embryonic gene (ζ) and two adult genes (α1 and α2). The β-like genes comprise an embryonic gene (ε), duplicated fetal (γ) and adult (β and δ) genes. Expression of these genes is developmentally regulated during fetal life. The array of α- and β-like genes on their respective chromosomes reflects the order in which they are expressed. The control of the switch from embryonic, to fetal and then to adult globin synthesis, seems to be controlled by the interaction between several transcription factors, the locus control regions upstream of the gene cluster and the promoter regions of the individual genes. FIGURE 29.3 Organization of the human globin gene cluster and developmental changes in haemoglobin. Normally, the synthesis of α- and β-like chains is carefully balanced to avoid the accumulation of free globin chains, although it is not clear how this balance is maintained. In the thalassaemias, where this balance is severely disturbed, the excess of one type of globin chain is central to the pathophysiology. The switch in globin synthesis during development has important implications for clinical expression of the haemoglobinopathies. Complete failure of α-chain synthesis, which, since the α genes are duplicated on each chromosome, occurs only when there is loss of function involving all four α genes, becomes evident early in fetal life. By contrast, even if there is little or no normal β-chain synthesis, the effects will not be manifest until the switch from fetal γ to adult β gene expression is completed after birth. This switch is gradual, and abnormalities of β globin synthesis rarely result in clinical problems before three months of age. Reversing this switch, and reactivating fetal haemoglobin is also potentially curative for diseases caused by mutations of the β globin genes. The thalassaemias are one of the commonest human autosomal recessive disorders, with approximately 120 000 severely affected individuals born annually worldwide. The most common and clinically significant forms are α and β thalassaemia and the β thalassaemia-like structural variant, haemoglobin E. These may be further subdivided according to whether there is no output of globin (α0 or β0 thalassaemia) or some preservation of globin chain synthesis (α+ and β+ thalassaemia). In the case of α thalassaemia, this is complicated further by the duplication of expressed α genes. α Thalassaemia is clinically significant only when three or four α globin genes are lost. The clinical consequences of thalassaemia in the homozygous (or compound heterozygous) state may be understood in terms of the imbalance in globin chain synthesis that results from absent or reduced synthesis of either α or β globin. Unpaired α or β globin chains are toxic, and damage the developing red cell, causing it to die whilst still in the marrow, which is characteristic of thalassaemia and called ineffective erythropoiesis. Heterozygotes are unaffected clinically, and manifest the condition as slight anaemia with reduced red cell size and haemoglobin content. Increasing globin chain imbalance, as occurs in homozygous and compound heterozygous states, results in more marked anaemia and bone marrow expansion, with corresponding development of symptoms. A characteristic of thalassaemia syndromes is their marked phenotypic heterogeneity. For over 20 years now, the molecular basis of thalassaemia has been studied and understood in great detail. The number of mutations identified as causing thalassaemia is large and continues to grow. However, in most populations, a small range of 5–10 different thalassaemia mutations accounts for about 90% of the mutant alleles found in that particular area. Though particularly common in South-East Asia, where carrier rates may reach 50%, α thalassaemia is widely distributed in all major populations apart from northern Europeans (see Fig. 29.1). The pathophysiological effects of α thalassaemia reflect the degree of impairment in α globin production (see Table 29.1). The majority of cases are due to deletions involving one or both α globin genes. TABLE 29.1 The α thalassaemia syndromes a − α, denotes loss of function of one α gene on a chromosome, i.e. α + thalassaemia. Haemoglobin Bart’s hydrops fetalis is the most severe form of α thalassaemia, where all four α genes are affected, abolishing or severely diminished α chain synthesis. The disease becomes manifest in fetal life. With the failure of normal production of fetal haemoglobin (α2 γ2), surplus fetal γ-chains combine to form tetramers (γ4) recognized electrophoretically as haemoglobin Bart’s. The fetus is only able to survive at all in utero because of the presence of increased amounts of the embryonic haemoglobin Hb Portland (ζ2γ2). Haemoglobin Bart’s possesses markedly increased oxygen affinity and is ineffective as an oxygen transporter. In an attempt to compensate for impaired tissue oxygen delivery, there is erythroblastosis with extramedullary haemopoiesis, leading to hepatic and splenic enlargement. Severe functional anaemia leads to tissue hypoxia, increased capillary permeability, and cardiac failure, which lead ultimately to fetal hydrops and death in utero or stillbirth in late pregnancy. Haemoglobin H disease occurs when there is loss of function of three out of four α genes. There is sufficient residual α-chain synthesis to allow production of some normal fetal and adult haemoglobin, and fetal development is generally normal. A variable amount (10–40%) of Hb Bart’s is detectable at birth. After birth, the excess β-chains form tetramers detectable electrophoretically as the fast variant haemoglobin H (β4). Haemoglobin H (HbH) precipitates within red cells with the formation of inclusion bodies leading to shortened red cell survival. Staining of these inclusions with the redox dye brilliant cresyl blue serves as a useful diagnostic test. The clinical effects of HbH disease vary considerably. Most patients have a mild to moderate haemolytic anaemia, which may be exacerbated during pregnancy or parvovirus B19 infection, accompanied by splenomegaly. Growth and development are usually normal. As in other congenital haemolytic anaemias, there is an increased tendency to form pigment gallstones. It follows that Hb Bart’s hydrops fetalis and HbH disease occur only when at least one parent carries the α0 genotype (–/αα). This provides an explanation for the observation that these disorders occur primarily in southern China, South-East Asia and the eastern Mediterranean where α0 thalassaemia is prevalent, and are not seen in other parts of the world, for example Africa and India, where α0 thalassaemia is very rare. Individuals in whom one or two α genes are dysfunctional are clinically unaffected, although they show thalassaemic red cell indices and traces of Hb Bart’s at birth (see Table 29.1). This form of α thalassaemia trait is very common in most populations, with a 3.7 kb deletion accounting for most cases. Non-deletional forms of α thalassaemia are increasingly recognized as DNA analysis becomes more widespread and sophisticated. β Thalassaemia is prevalent throughout tropical and North Africa, the Mediterranean, Middle East and large parts of south and South-East Asia, including India (see Fig. 29.1). In some parts of the world, for example Cyprus, the heterozygote (carrier) frequency may reach 15%. Over 300 different mutations of the β globin gene or its promoter are known to cause β thalassaemia, the majority being single nucleotide substitutions (point mutations) or small deletions. Geographically, these segregate non-randomly, so that four or five specific mutations account for the majority of β thalassaemia genes in individual ethnic or geographic groups. Each mutation typically occurs on a single β globin gene haplotype, suggesting that in most instances individual β thalassaemia mutations have arisen historically on a single occasion.
The haemoglobinopathies
INTRODUCTION
The structure and function of haemoglobin
The genetic control of haemoglobin synthesis
THE THALASSAEMIAS
α Thalassaemia
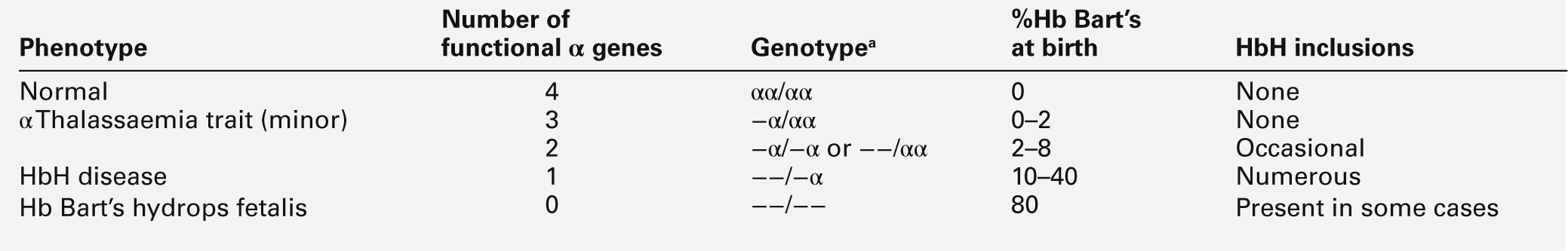
−−, denotes loss of function of both α genes on a chromosome, i.e. α0 thalassaemia.
β Thalassaemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


