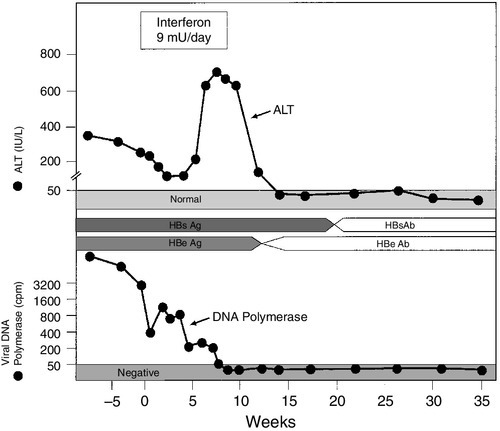
CHAPTER 14 Adrian Bomford; Roy A. Sherwood CHAPTER OUTLINE CLASSIFICATION OF LIVER DISEASE ACUTE HEPATITIS AND ITS SEQUELAE Laboratory criteria for liver transplantation Differential diagnosis of chronic hepatitis PRIMARY SCLEROSING CHOLANGITIS Liver pathology in alcoholic liver disease Use of laboratory tests in clinical practice Non-alcoholic fatty liver disease Vascular disturbances in cirrhosis Sex hormones and their binding proteins NEOPLASTIC DISEASE OF THE LIVER AND BILIARY TRACT Hepatocellular carcinoma and α-fetoprotein INHERITED METABOLIC DISORDERS INVOLVING THE LIVER Iron overload and hereditary haemochromatosis Other inherited metabolic diseases The immediate postoperative period Liver disease is best classified according to its presumed aetiology (Box 14.1). This is then qualified by the pathological state of the liver where known or inferred: usually cirrhosis, inflammation (i.e. hepatitis) or cholestasis. Thus, terms such as alcoholic cirrhosis, viral hepatitis or cholestasis of pregnancy all conform to this classification. Some idea of the severity of the disease is given by the terms ‘compensated’ and ‘decompensated’. The term ‘compensated’ implies that, although the liver has been damaged it can, because of its large functional reserve, still function adequately. On the other hand, the term ‘decompensated’ implies that the liver is failing in some vital function. The terms are not precisely defined, but the development of ascites with or without peripheral oedema, jaundice or hepatic encephalopathy, in a patient known to have liver disease, would all be regarded as signs of decompensation. Finally, the presumed evolution of the disease in time is given by the terms fulminant, acute, subacute or chronic. In chronic liver disease, additional terminology often employed to provide an indication of severity, especially when assessing histological changes in the liver, includes the terms active or inactive and mild, moderate or severe. The term ‘hepatitis’ indicates that there is hepatic inflammation, for which there are many causes: viral infections, drugs and toxins (including alcohol) are common, while autoimmune causes are infrequently encountered in the primary care setting, but in a tertiary centre make up a substantial proportion of patients. The standard ‘liver function tests’ (LFTs) are useful to determine that hepatitis is present, how severe it is, to document the progression of the disease and to assess the response to therapy. However, their role in identifying the aetiology of the inflammation is limited. When the patient presents with an acute hepatitis, the liver function tests characteristically show the so-called ‘hepatitic’ picture: • a modest (less than twice the upper limit of the reference range) increase in plasma alkaline phosphatase (ALP) activity • bilirubinuria and, in more severe cases, hyperbilirubinaemia, which is clinically detectable as jaundice when the total plasma bilirubin rises to > 40–50 μmol/L. As a broad generalization, plasma activities of the aminotransferases reflect the severity of the disease. Certainly, patients who progress to fulminant hepatitis usually have exceptionally high plasma activities, with elevations of 20–40 times the upper reference limit. However, the relationship is not strictly quantitative, for lower activities (2–3 times the upper reference limit) can be found in individuals with severe hepatic necroinflammatory activity on histological examination of liver biopsy material. There are two aspects to this part of the diagnostic process. The first is to distinguish between viral hepatitis and other, non-viral, causes of a hepatitic illness and then, within each group, to identify the causative agent. Generally speaking, the standard LFTs are not helpful in separating viral from non-viral causes, and further serological testing, radiological imaging or histological examination will be required. Plasma aminotransferase activities are not usually grossly raised in alcoholic hepatitis; in this condition, a ratio of AST:ALT of > 2 is characteristic, while in hepatitis due to other agents the ratio is usually < 2. In fulminant hepatitis, aminotransferase activities may be close to the reference ranges by the time the patient reaches hospital, emphasizing that results of LFTs can change rapidly with time and depend on the particular clinical course that ensues. Generally speaking, acute infection with hepatitis B virus is more severe than that caused by hepatitis A, and biochemical abnormalities are more protracted. Raised plasma IgM concentration and atypical lymphocytes are more characteristic of type A, but the standard LFTs do not permit distinction between the different types of viral hepatitis. The specific diagnosis is established by serological tests for the hepatitis viruses (Table 14.1). The extent to which individual patients require biochemical and serological investigation depends on the clinical situation. For example, in children, a clinical diagnosis can be established with reasonable certainty if the clinical features are compatible with hepatitis A (infectious hepatitis), particularly if the physician knows of local occurrence. In adults, particularly where there has been no contact with infectious hepatitis, LFTs should be performed together with appropriate serology. After a variable incubation period (the duration of which depends on the type of virus and the viral load), there is usually a rise in plasma aminotransferase activities, although some infants can be infected without any disturbance of LFTs. In many cases, particularly among young children, an elevated AST or ALT is the only indication of the hepatitic process and the patient remains asymptomatic. When symptoms do develop, they are coincident with the maximal aminotransferase activities. Taking infection with the hepatitis A virus as an example, the patient begins to feel unwell and lethargic and may become anorexic, with transient diarrhoea being frequent. Pyrexia can develop and the liver is found to be enlarged and tender. The urine may become dark (owing to bilirubinuria), the stools pale, and in more severe cases, jaundice becomes evident a few days later. Symptoms tend to resolve with the onset of jaundice, which usually subsides over a few days. Following infection with this virus, the patient may, despite being asymptomatic, enter a cholestatic phase. This is heralded by rising plasma activities of ALP and γ-glutamyltransferase (γGT) that can persist for many weeks and may cause considerable diagnostic confusion. Particularly in the older patient, it is an indication for ultrasound examination to rule out obstruction of the biliary tract. The jaundice usually begins to resolve before aminotransferase activities return to the reference range, but it has been a frequent observation that bilirubin may disappear from the urine while the patient remains clinically jaundiced. The explanation for this situation lies in the development of ‘bili-albumin’ (see p. 239). There are three main possible outcomes of acute viral hepatitis, each with its own characteristic progression of clinical features and pattern of LFTs. As noted above, the plasma aminotransferase activities start to rise before the onset of jaundice and often before hepatomegaly is clinically detectable. Activities begin to fall coincidentally with the onset of jaundice and symptoms, and return to normal (AST before ALT) at the same time as, or shortly before, the plasma bilirubin concentration. This is the natural history in the great majority of patients and, unless jaundice develops, many patients will be unaware that they have had an acute hepatitis. This mode of progression is limited to viral hepatitis types B and C, but a relapsing course for hepatitis A is well described and there are occasional reports of type A triggering autoimmune hepatitis in susceptible individuals. Persistence of symptoms, signs and/or abnormal liver tests (particularly aminotransferase activity in the range of 2–10 times the upper reference limit) for more than six months constitutes, by definition, chronic hepatitis (see below). The accompanying changes in viral serology are complex and will not be considered in the present discussion. Very rarely (in < 1% of patients), any type of viral hepatitis may pursue a fulminant course. This course of events is characterized by grossly elevated plasma aminotransferase activities, an increasingly prolonged prothrombin time (PT) or international normalized ratio (INR) and the development of hepatic encephalopathy, which may occasionally develop before jaundice becomes a prominent feature. The prognosis is poor. The syndrome of acute liver failure (ALF), one cause of which is acute viral hepatitis, is described in more detail below. Acute liver failure implies the development of severe hepatic dysfunction within six months of the first onset of liver disease and in the absence of any pre-existing liver disease. Hepatic encephalopathy and a prolonged and persistent increase in PT are the characteristic features, and where these occur within eight weeks of the first symptom, the condition is known as fulminant hepatic failure (FHF). In those who survive, there are no long-term hepatic sequelae. The commonest cause in the UK is paracetamol overdose (Chapter 40) followed by viral hepatitis types B and E; ALF due to hepatitis C virus is extremely rare. Rarer causes include adverse drug reactions, other viruses, Amanita mushroom poisoning, Wilson disease, secondary hepatic malignancy, lymphoma and recreational drugs. Laboratory investigation is aimed at determining the cause of the ALF as well as assessing its consequences and complications (which are very similar irrespective of the aetiology). Measurement of the blood concentration of paracetamol is important, particularly in view of the beneficial effect of treatment by N-acetylcysteine infusion. The serology of viral causes is complicated because the hepatitis B surface antigen (HBsAg) may become undetectable before the patient reaches hospital. In general, with ALF due to causes other than paracetamol overdose, it is usually too late for specific treatment for the condition (e.g. D-penicillamine for Wilson disease) to be effective and management is by general supportive measures and, if feasible, liver transplantation. The standard LFTs show a grossly hepatitic picture at the time of onset of the encephalopathy. Jaundice is deep and progressive and plasma aminotransferase activities of several thousand U/L are usually found, although these may have fallen considerably by the time the patient is sent to a referral centre. A rare cause of FHF is massive metastatic tumour infiltration. This is characterized by hepatomegaly (in contrast to the reduced-size liver found with most other causes of FHF) and a much more cholestatic picture – such that the AST:ALP ratio is < 4 rather than the converse, as in most other cases. Coagulation defects are a consistent finding. Plasma concentrations of fibrinogen are decreased as are those of factors II, V, VII, IX and X. These are reflected in a prolonged PT or INR which, in the UK, is used as the main measure of severity of disease and is the most widely used parameter for following progress. Hypoglycaemia, owing to a combination of impaired gluconeogenesis and glycogen breakdown and synthesis, is such a consistent finding that glucose infusion is a routine part of management, particularly during transfer to a referral centre. Where paracetamol is involved, it may interfere with blood glucose estimation, resulting in erroneously high values. Acute kidney injury is a frequent and ominous complication that occurs in at least 50% of patients with FHF, particularly with paracetamol poisoning. In about one-half of patients, the renal lesion is acute tubular necrosis, and in the remainder it is ‘functional’ renal injury (see p. 261). Currently, with the best supportive care, 30–50% of patients with fulminant hepatic failure will survive; the figures are rather better for those with paracetamol overdose and rather poorer for those with viral hepatitis, particularly those classified as non-A–E, or drug reactions. This poor prognosis has led to the introduction of liver transplantation as a therapeutic option. It is clearly important to reserve this option for those with the worst prognosis and who are most unlikely to recover with supportive treatment. With this in mind, a number of laboratory-based criteria have been identified to ascertain the likelihood of death. The major adverse factors are an arterial hydrogen ion concentration > 50 nmol/L (pH < 7.3), and progression to grade III coma (see p. 259) with an INR of > 10 and plasma creatinine concentration > 300 μmol/L. Recently, there has been considerable interest in the measurement of plasma factor V concentration. If the ratio of factor V to factor VIII is very low, the chance of survival without transplantation is very small. Chronic hepatitis is usually defined as a condition in which clinical or biochemical features of liver disease persist for more than six months. However, it has become clear that many patients can have the condition for much longer periods before it manifests itself in this way. Indeed, occasionally, such individuals may present with what at first appears to be an acute hepatitis that, on further investigation, proves to be an acute exacerbation of a previously asymptomatic chronic process (see below). On histological grounds, the condition was previously classified into two categories: chronic persistent hepatitis (CPH), defined as inflammation confined to the portal tracts with no necrosis of hepatocytes, and chronic active hepatitis (CAH) characterized by inflammatory cells spilling out into the hepatic parenchyma and damaging periportal hepatocytes in a hallmark pattern described as ‘piecemeal necrosis’. Chronic persistent hepatitis was believed to be a benign condition of little clinical significance, with a good prognosis. Chronic active hepatitis, in contrast, was envisaged as a disorder with a more serious prognosis, which frequently progressed to cirrhosis, with the inflammatory activity often continuing even after cirrhosis developed (so-called ‘active cirrhosis’). Although CPH and CAH were purely histological descriptions of the morphological changes seen in patients with chronic liver disease, they were embraced by hepatologists and gastroenterologists as clinical entities. However, during the 1970s and 1980s, it was increasingly recognized that the associated changes could be found in patients with several quite distinct causes of chronic hepatitis (Table 14.2). Somewhat surprisingly, rather than questioning the validity of CPH and CAH as distinct syndromes, this led to expansion of the list of diseases considered to be capable of causing them. By the 1990s, the previously accepted view that CPH was associated with a good prognosis was being challenged, because it became apparent that CPH and CAH are changes at either end of a spectrum of morphological changes seen in chronic hepatitis, and that transitions between them can and do occur during flares of disease activity and periods of remission, regardless of the aetiology of the process. TABLE 14.2 Differential diagnosis of chronic hepatitis a For explanation, see α1-Antitrypsin deficiency on p. 268. ANA, antinuclear antibody; LKM, liver-kidney microsomal antibody; MCV, mean corpuscular volume; SMA, smooth muscle antibody. Other criteria for CPH and CAH were also found to be untenable. A requirement for markedly elevated plasma aminotransferase activities for diagnosis of CAH could not be upheld because it was recognized that these enzymes are poor correlates of histologically assessed activity in chronic liver disease, and that mild elevations of aminotransferases do not exclude severe disease. The temporal criterion for duration of disease of at least six months (to distinguish chronic from acute liver disease) also proved difficult to apply because it is often not possible to define the time of onset and, as noted above, patients presenting with acute hepatitis who had clear evidence of chronic liver disease were being identified. Current recommendations for defining and describing chronic hepatitis are that the terms CAH and CPH should be abandoned in favour of precise morphological descriptions, graded for necroinflammatory activity and staged for degree of fibrosis. While the term ‘piecemeal necrosis’ may be retained, the terms ‘periportal hepatitis’ or (preferably) ‘interface hepatitis’ should be used to describe the changes previously associated with CAH. Description of the changes previously associated with CPH should employ terms such as mild or moderate portal or periportal hepatitis (as appropriate) without significant necrosis. All of these terms should be qualified by aetiological designations (e.g. autoimmune hepatitis, chronic hepatitis B, C, D etc.), wherever possible and practicable. Laboratory investigation plays a crucial role in the differential diagnosis and management of chronic hepatitis (see Table 14.2), although histological examination of liver biopsy material is usually required to assist in assessing severity and providing additional aetiological information. Typically, chronicity is defined as persistence of a hepatitic pattern of abnormal LFTs over several months. The plasma aminotransferase activities are usually elevated to 2–10 times the upper reference limits. Plasma alkaline phosphatase is often normal or only slightly raised, although higher values may be seen in patients where cirrhosis has developed with associated distortion of the hepatic architecture. Plasma bilirubin concentration is often also normal or mildly elevated but, in severe cases, there can be a profound hyperbilirubinaemia with jaundice. The PT/INR and other markers of coagulation are often mildly abnormal, and the plasma albumin concentration may be at the lower end of the normal range, reflecting decreased hepatic synthetic function. Marked hypoalbuminaemia is, however, a late feature associated with advanced cirrhosis. Depending on the aetiology of the chronic hepatitis, plasma immunoglobulin concentrations may also be elevated, sometimes quite markedly (see below). The above abnormalities may be a sequel to an acute hepatitis, in which case the aminotransferase activities may have risen to 10–20 times the upper reference limits before falling back to the somewhat lower values more characteristic of chronic progression. However, a clinically evident acute phase may not have been apparent especially if the patient did not develop jaundice. Indeed, chronic hepatitis is not infrequently diagnosed incidentally during routine health screening or investigation of some other condition. Notably, profound fatigue is a common feature of all forms of chronic hepatitis, and the latter should, therefore, be considered in the differential diagnosis of chronic fatigue syndromes. The specific diagnosis is based on virological tests. The detection in serum of the hepatitis B surface antigen (HBsAg) indicates that the patient is infected with the hepatitis B virus (HBV). Clinical information and the results of LFTs can be useful in deciding whether the infection is acute, but this can be confirmed by the appearance of IgM antibodies against the HBV core antigen (HBc). The gradual disappearance of HBsAg, with sequential detection of IgG antibodies to HBc and antibodies to surface antigen (HBsAb) indicates clearance of the virus, an event that occurs in the majority of adults (90%) acutely infected with it. Other markers of viraemia are derived from different components of the virus, including hepatitis B ‘e’ antigen (HBeAg) and HBV DNA, and these disappear with viral clearance. The decline in HBe antigenaemia is accompanied by the appearance of antibodies to the ‘e’ antigen (HBeAb) and is known as seroconversion. In contrast to infection acquired in adulthood, perinatal infection becomes chronic in 95% of patients, with the virus persisting into adult life. Chronic infection is associated with a number of outcomes. Many individuals are asymptomatic, with the virus detected incidentally and a serological profile in which HBsAg and HBeAb are detected and HbeAg is negative. Although such people frequently have normal liver function tests, and have long been termed ‘healthy carriers’ and been considered to be of low infectivity, it has become clear that a significant proportion have marked histological damage on liver biopsy and detectable circulating HBV DNA. Others have clear evidence of cirrhosis and, in such individuals, there is a 5% annual risk of developing hepatocellular carcinoma. Hepatitis C virus (HCV) infections are identified initially by detection of antibodies (anti-HCV) to the virus and chronic infections confirmed by seropositivity for the viral RNA (HCV-RNA). Antiviral therapy for chronic hepatitis B and C, using interferons, with or without other antiviral agents to inhibit viral replication, is now well established, and comprehensive guidelines for treatment exist. Thus, tests for genomic material of both viruses (HBV-DNA and HCV-RNA, respectively) are usually performed, initially to assess the viral load before treatment and subsequently to monitor the patient’s response to treatment. Plasma aminotransferase activities are also usually monitored as an adjunct to the above, since they are cheap to perform on a frequent basis and a return to normal values suggests at least some response to treatment. In the case of HBV, successful treatment is heralded by a change from HBeAg to HBeAb positivity and this event is accompanied by an acute hepatitic illness, the so-called ‘hepatitic flare’ (Fig. 14.1). Hypothyroidism is a side effect of interferon therapy, particularly in patients with chronic hepatitis C, and thyroid function tests should be monitored before treatment and at 3-monthly intervals. FIGURE 14.1 Changes in ALT activity in a patient with chronic hepatitis B virus infection successfully treated with interferon. Note the hepatitis response heralding clearance of HBsAg and appearance of the antibody. Clearance of the virus is also indicated by a decrease in viral DNA polymerase activity. Procedures for the handling of potentially dangerous and infectious samples are not the concern of this chapter, but it is worth noting that as many as 1–2% of inner city populations may be carriers of either HBV or HCV. The majority of these will have no symptoms of liver disease and their blood samples may be referred to the laboratory for causes unrelated to liver disease. Caution should therefore be exercised in the handling of all blood products in view of the possibility of occult hepatitis viral infections. In perhaps 10% of patients with alcoholic liver disease, the histological features of interface hepatitis, similar to those in other forms of moderate/severe chronic hepatitis can be observed. There is evidence that some such cases may be related to coexistent hepatitis C virus infection or other underlying causes. The problem that this presents for differential diagnosis is discussed in detail under the heading of alcoholic liver disease. Chronic hepatitis is a rare presentation of this rare disease (see p. 267), but is of considerable importance because treatment can be instituted and further liver damage avoided. Generally, the plasma activities of aminotransferases are low in comparison with the extent of histological activity. The diagnostic work-up is considered in detail below, but the initial screening procedures include measurement of plasma copper and caeruloplasmin concentrations and ocular slit-lamp examination for Kayser–Fleischer rings. The diagnosis must be considered in all patients with chronic hepatitis, particularly those in the younger age groups. This is another rare cause of chronic hepatitis (see p. 266). Plasma α1-antitrypsin concentrations are usually low but, occasionally, because of active inflammation, they may be within the reference range. It is therefore essential that the α1-antitrypsin phenotype is determined, particularly in younger patients. This is a rare cause of chronic hepatitis. However, it is one of the few causes of chronic liver disease in which drug treatment is highly effective in the great majority of patients, and accurate diagnosis is essential. This is established partly by careful exclusion of the other causes listed above and partly by the finding of a suggestive pattern of biochemical, immunological and histological abnormalities. The biochemical liver tests show a hepatitic pattern (see above), but plasma aminotransferase activities are often only moderately elevated and bilirubin concentrations are frequently normal. Typically, there is a marked hypergammaglobulinaemia with selective elevation of the IgG concentration (which can be as high as 100 g/L). Approximately 80% of AIH patients will also have significant titres (> 1:40) of organ non-specific autoantibodies (smooth muscle antibody (SMA), antinuclear antibody (ANA) or anti-liver-kidney microsomal (LKM) antibodies). Standard treatment involves immunosuppressive therapy with prednisolone and the steroid-sparing agent azathioprine. Plasma aminotransferase activities do not correlate well with histologically assessed disease activity in AIH. However, such tests, together with plasma IgG concentrations, are cheap and can be repeated more easily than liver biopsy, so are used routinely to monitor response to immunosuppressive therapy. Some 80–95% of AIH patients respond to the standard therapy of prednisolone and azathioprine, with aminotransferase activities and IgG concentrations falling to at least 50% below their initial values within three months. Dosages are titrated against the aminotransferase and IgG values, being progressively reduced to maintenance doses as these parameters fall towards the normal range that, in most patients, is achieved within one year of starting treatment. However, this ‘biochemical remission’ does not represent a complete response. It usually requires at least a further year of treatment before complete histological remission is achieved. Current recommendations are that treatment should be continued for at least two years (preferably four), with regular monitoring of aminotransferase activities and IgG concentrations; before any attempt is made to withdraw treatment, histological remission should be confirmed. Withdrawal of all immunosuppression is rarely achieved, possibly because it is not routinely attempted, but gradual reduction in the dose of prednisolone to very low levels, or complete withdrawal, is feasible so that immunosuppression can be maintained with azathioprine alone. If complete withdrawal of immunosuppressive treatment is achieved, the patient should be monitored indefinitely, because even a complete histological response does not represent a true ‘cure’ and there is a lifelong risk of relapse of the disease. The latter is heralded by a rise in aminotransferase activities and IgG concentrations. Importantly, interpretation of biochemical test results should not focus solely on the upper reference limits for the various parameters but should take account of what may be normal for the individual patient. By definition, half of the normal population will have values below the geometric means of the reference ranges and, for such individuals, a rise in a parameter to (or just above) the upper reference limit may represent a doubling of their normal value. This is a chronic cholestatic condition of unknown cause in which there is destruction of septal and interlobular bile ducts. Females are affected far more frequently than males. Most patients present with pruritus, jaundice or non-specific symptoms including tiredness and hepatic pain. However, an increasing number are detected incidentally when abnormal liver tests, particularly an increased activity of alkaline phosphatase, are detected during opportunistic screening. Although PBC is considered to be an autoimmune liver disease, and twin and family studies suggest that there is a significant genetic component, it is clearly a complex disease and little progress has been made in defining the genes responsible. Biochemical liver function tests reveal a characteristic cholestatic picture with, as the disease progresses, increasing plasma alkaline phosphatase (ALP) (of biliary origin), increasing bilirubin concentration and falling albumin. The plasma concentrations of both pentameric and monomeric IgM are frequently raised (cf. AIH). The most specific serological tests are those for antimitochondrial antibodies (AMA), which are detectable in about 95% of patients. Antimitochondrial antibodies recognize several distinct autoantigens, but the most characteristic for PBC are the M2 antibodies that react with epitopes on the E2 components of the pyruvate dehydrogenase complex (PDC-E2), the branched chain oxoacid dehydrogenase (BCOADH-E2) and the oxoglutarate dehydrogenase complexes. Laboratory tests, particularly the plasma bilirubin concentration, are used in assessing prognosis (see p. 248). This is a progressive disease characterized by diffuse inflammation and fibrosis of the extrahepatic and/or intrahepatic biliary system, which leads to obliteration of the intrahepatic ducts and, eventually, biliary cirrhosis (see below). It is diagnosed on the basis of characteristic cholangiographic appearances together with compatible clinical, biochemical and histological features, and exclusion of several other conditions known to cause secondary sclerosing cholangitis, such as biliary calculi and previous biliary tract surgery. Men are affected more often than women and, again, although considered an autoimmune disease, the disease is genetically complex and remains poorly characterized. With the advent of endoscopic retrograde pancreatography (ERCP), PSC began to be diagnosed earlier in its natural history, particularly following the initial finding of persistently abnormal LFTs in patients with inflammatory bowel disease, which coexists with PSC in more than 50% of cases. In the early stages, there is usually a minor elevation of plasma aminotransferase activities and moderate elevation of ALP, which progresses, over several years, to a very severe cholestatic condition with deep jaundice. By this stage, plasma ALP activity often reaches more than ten times the upper reference limit. Death from liver failure ultimately results unless a liver transplant can be performed. However, the disease has been known to recur in the grafted liver. Rapid clinical deterioration with a progressive cholestatic picture may indicate the development of cholangiocarcinoma, a complication with a lifetime risk of 10–15% in this condition. CA19-9 concentrations may be elevated when a cholangiocarcinoma has developed but this is not a specific finding as CA19-9 is also increased in other cholestatic liver diseases. Copper retention appears to be a general feature of chronic cholestatic conditions: in PSC most patients will have raised hepatic copper concentrations (of a similar order to that seen in Wilson disease and primary biliary cirrhosis) and urinary copper concentrations are typically raised. However, in contrast to Wilson disease, the plasma caeruloplasmin and copper concentrations will remain within the reference range (see p. 267). Excessive alcohol (ethanol) consumption is by far the commonest cause of liver disease in the Western world, although it often exists as part of a wider spectrum of social, psychological and pathological effects of alcohol-related damage. Most current evidence suggests that alcohol itself is the primary cause of the liver damage, although associated malnutrition may be a contributory factor. Considering that there is an enormous variation in the amount of alcohol drunk by different individuals, the time course over which it is consumed and individual susceptibility to tissue injury, it is not surprising that the adverse effects on the liver vary widely. Alcohol is absorbed from the stomach and small bowel. Absorption is most efficient when there is no other food (particularly carbohydrate) in the gut and when its concentration in the ingested fluid is of the order of 20%.
Acute and chronic liver disease
CLASSIFICATION OF LIVER DISEASE
ACUTE HEPATITIS AND ITS SEQUELAE
Differential diagnosis
Acute viral hepatitis
Outcome of acute viral hepatitis
Complete resolution
Progression to chronic liver disease
Progression to acute liver failure
ACUTE LIVER FAILURE
Laboratory features
Laboratory criteria for liver transplantation
CHRONIC HEPATITIS
Type
Laboratory tests
Comments
Viral
type B
HBsAg
type C
Anti-HCV; HCV-RNA by PCR
Has characteristic histological features
Alcoholic
Blood alcohol, γGT, MCV, desialylated transferrin
Laboratory tests are supplementary to clinical history
Drugs
May develop autoantibodies
Oxyphenisatin, methyldopa, isoniazid, dantrolene etc.
α1-Antitrypsin (AT) deficiency
Low α1-AT concentration; typical phenotype (PiZZa)
Characteristic eosinophilic globules on histology
Wilson disease
Low caeruloplasmin, high tissue copper and urinary copper concentration
Low ALP activity; low AST/ALT activity for degree of inflammation
Autoimmune
type 1
Anti-SMA and/or ANA
Markedly raised γ-globulin and IgG
type 2
Anti-LKM antibodies
Differential diagnosis of chronic hepatitis
Viral hepatitis types B and C
Alcohol
Wilson disease
α1-Antitrypsin deficiency
Autoimmune hepatitis (AIH)
Monitoring response to therapy
PRIMARY BILIARY CIRRHOSIS (PBC)
PRIMARY SCLEROSING CHOLANGITIS (PSC)
ALCOHOLIC LIVER DISEASE
Ethanol metabolism
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


