Substance
Bruto formula
Molecular mass
1 mg substance = base (mg)
Codeine (pure substance)
C18H21NO3
299.4
1
Codeine. H2O (Ph. Eur.)
C18H21NO3.H2O
317.4
0.94
Codeine phosphate.½H 2 O (Ph. Eur.)
C 18 H 24 NO 7 P.½H 2 O
406.4
0.74
Codeine phosphate. 1½ H2O (Ph. Eur.)
C18H24NO7P.1½H2O
424.4
0.71
Codeine HCl.2H2O (BP)
C18H21NO3Cl.2H2O
371.9
0.81
In addition to water of crystallisation, where the water molecules are in stoichiometric proportion to the substance, water also appears as adsorbed water, such as in the case of prednisolone disodium phosphate.
23.1.5 Salt-and Ester Forms
Many substances appear either as unconjugated or as salt or ester form. If the substance is a salt or an ester of the substance to be processed, it will be seen from the dosage specified whether or not there is a need for a conversion factor to be applied.
Exchanging a base/acid with the corresponding salt, or an alcohol/acid with an ester almost always has biopharmaceutical, pharmacokinetic or pharmacodynamic consequences of clinical importance. Sometimes these are desirable, but most often they are not. Be guided by the biopharmaceutical information in reference books (such as Martindale [3]) or the composition of the licensed medicine. Some examples:
23.1.5.1 Corticosteroids
The esters of monovalent fatty acids (triamcinolone acetonide) are usually used in cutaneous preparations. Free corticosteroids can usually be administered orally. When a solution in water is needed the esters of a multivalent acid in the salt form (-disodium phosphate, disodium succinate) should be used. This also applies to processing corticosteroids in a lipid based suppository.
23.1.5.2 Excipients
It is also important, with regard to excipients as to what chemical form is used. DL-alfa-Tocopherol acts as antioxidant in an alcohol-water mixture; the esters have no effect.
23.1.5.3 Label Claims
In many cases the labelling of the strength indication is clear, but errors can easily occur. Some examples to illustrate how label claims should be interpreted:
Garamycin® ampoule: according to the label it contains 10 mg per mL gentamicin base as sulfate. Thus the ampoule contains gentamicin sulfate and the indication of the strength relates to gentamycin ion.
Erythrocin® -suspension and tablets: according to the label the content is related to erythromycin base.
‘Ritalin capsules’; when these are to be prepared from methylphenidate hydrochloride it is simple, since the strength of Ritalin® tablets relates to methylphenidate hydrochloride.
Bethamethason cream: in this case however there is firstly the choice between valerate and dipropionate, each of which represents a different corticosteroid activity class. Then in both cases the desired concentration in relation to the amount of free betamethasone has to be calculated. The strength of the authorised products is specified respectively as valerate or dipropionate.
Iodinated povidone is a colloidal complex and problems can arise with the content indication of iodinated povidone solution. Betadine® contains 10 % iodinated povidone complex, corresponding to a concentration of approximately 1 % iodine.
Neomycin: Still more unclear is the issue of the interpretation of label claims with neomycin sulfate. Neomycin sulfate Ph. Eur. is “a mixture of sulfates or substances”. The sulfate content may vary between 27 % and 31 % calculated on the dried substance and the water content can be up to 8 %. It is therefore not possible to derive an exact conversion factor when a neomycin preparation is prescribed. In practice no difference is made between prescribing neomycin or neomycin sulfate, as neomycin as such is not available and the dose cannot be based on experience with that substance. So it turns out from the context that there is no need for conversion.
An important point can be the difference in application of certain salts. An example is chlorhexidine digluconate, that is available as 20 % solution (Hibitane®). Chlorhexidine chloride is not water-soluble and is available as dry powder (under the same brand name Hibitane®). If in the doctor’s request the brand name is used, it will be seen from the context what salt is intended and, in the case of the gluconate, whether the strength indication relates to the digluconate as such or to the 20 % solution. To complicate the matter even more, a third variant is available, i.e. chlorhexidine acetate that is insoluble in water but soluble in propylene glycol.
23.1.6 International Units
International units are used for the strength indication of raw materials where this is not possible with units of mass. This will be the case, for example, if the raw material consists of a long chain with a varying number of functional groups, if the raw material consists of a mixture of substances with different mass per functional group or if for any other reason the mass is not strictly proportional to the strength of the raw material. Often the content of such substances must be determined with biological methods.
If a substance that is prescribed in international units (IU) needs to be weighed, the amount of substance is calculated with the conversion factor that is listed on the label or in the analytical report. For vitamins A and D the number of international units per gram is standardised. Other well-known examples are polymyxin and vitamin E. Prescribers often continue writing in units, even when, for the relevant active substance, a known chemical content has existed for a long time.
In some cases, the number of international units is indicated per packaging unit, for example, per ampoule of benzylpenicillin sodium. In that case, dilute the contents of the vial to a known volume and take back a calculated volume.
In Table 23.2 are some commonly used raw materials listed with, as far as is known, the equivalent amount of mass per unit.
Table 23.2
Substances of which the strength is expressed in IU
Substance | 1 IU = | 1 mg = | Remarks |
|---|---|---|---|
Bleomycin | 0.00067 mg (Ph. Eur.) | 1500 U (Ph. Eur.) | Clinical damage has been published [12] |
0.67–0.5 mg (USP) | 1.5–2 U (USP) | ||
Heparin | 0.0057 mg | 173 IU | Theoretical value |
Heparin calcium/sodium Ph. Eur. | For parenteral use: <0.0066 mg | ≥150 IU | 1. The exact amount of conversion should be mentioned on the label; |
2. Other standards apply for low molecular heparins; | |||
3. Values apply to dried substance; | |||
4. The tolerance in comparison to the declaration is 90–111 %; | |||
5. IU are not the same as USP units; However, the difference is not clinically relevant [4] | |||
Not for parenteral use: <0.0083 mg | ≥120 IU | ||
Neomycin sulfate Ph. Eur | <0.00154 mg | ≥680 IU | Value applies to dried substance |
Nystatin | Not oral: <0.227 microgram | ≥4400 IU | Value applies to dried substance |
Oral: <0.20 microgram | ≥5000 IU | ||
Polymixin B | 0.1 microgram | 10,000 IU | Theoretical value |
Polymixin B sulfate | 0.12 microgram | 8304 IU | Theoretical value |
Polymixin B sulfate Ph. Eur, commercially available | <0.167 microgram | ≥6000 IU | 1. Value applies to dried substance |
2. Ph. Eur. no longer applies IU | |||
Protamin sulfate | 0.01 IU heparin | 100 IU heparin | 1. Theoretical value |
2. Usually, the value is expressed in amount percentage | |||
Vitamin A as all-trans Retinol | 0.3 microgram | 3333 IU | 1. Theoretical values |
Vitamin A as all-trans Retinol palmitate | 0.55 microgram | 1818 IU | 2. The format in IU has already been left in 1956 |
Vitamin A as all-trans Retinol propionate | 0.36 microgram | 2786 IU | |
Vitamin A concentrate synthetic (oily form) Ph. Eur. (Vitaminum A densatum oleosum) | <0.002 mg | ≥500 IU | The format in IU has already been left in 1956 |
Vitamin A concentrate synthetic (powder form) Ph. Eur. (Vitaminum A pulvis) | <0.004 mg | ≥250 IU | |
Vitamin A concentrate, synthetic, solubilisate/emulsion Ph. Eur. (Vitaminum A in aqua dispergibile) | <0.01 mg | ≥100 IU | |
Vitamin D as Cholecalciferol (D3) or Ergocalciferol (D2) | 25 ng | 40,000 IU | Theoretical value |
Cholecalciferol concentrate (oily form) Ph. Eur. (Cholecalciferolum densatum oleosum) | <0.002 mg | ≥500 IU | |
Cholecalciferol concentrate (powder form) Ph. Eur. (Cholecalciferolum densatum pulvis) | <0.01 mg | ≥100 IU | |
Cholecalciferol concentrate (water-dispersible form) Ph. Eur. (Cholecalciferolum in aqua dispergibile) | <0.01 mg | ≥100 IU | |
Vitamin E as d-alfa-Tocopherol | 0.67 mg | 1.49 IU | The format in IU has already been left in 1956 |
Vitamin E as dl-alfa-Tocopherol | 0.91 mg | 1.1 IU | |
Vitamin E as d-alfa-Tocopheryl acetate | 0.74 mg | 1.36 IU | |
Vitamin E as dl-alfa-Tocopheryl acetate | 1.00 mg | 1.00 IU | |
Vitamin E as d-alfa-Tocopheryl hydrogen succinate | 0.83 mg | 1.21 IU | |
Vitamin E as dl-alfa-Tocopheryl hydrogen succinate | 1.12 mg | 0.89 IU | |
Vitamin E as d-alfa Tocopheryl polyethylenglycole-1000- succinate | 2.58 mg | 0.39 IU |
23.1.7 Microbiological Purity
23.1.7.1 Micro-organisms
For a number of raw materials Ph. Eur. has microbiological quality criteria for Total Aerobic Microbial Count (TAMC) and Total combined Yeast and Mould Count (TYMC) (see Sect. 19.6.3) and for absence of specific micro-organisms (see Sect. 19.6.4). Theoretically it would be logical to apply one requirement for all substances which are used in non-sterile preparations, but the approach should take into account the use of the final product. The General monograph Substances for Pharmaceutical use applies a general recommendation with regards to the microbiological contamination of the raw material and the microbiological requirements for the finished product. This contamination mainly refers to substances of vegetable, animal or mineral origin (see also Table 9.1 in Sect. 19.6.2). Even if such substances are not listed in the pharmacopoeia, it is recommended that microbiological contamination should be investigated; especially with herbs which can often be heavily contaminated. Examples of substances of which batches with questionable microbiological quality have been found are tragacanth gum, corn starch, kaolin and alginic acid [13].
Since vegetable raw materials with a sufficient microbiological quality are sometimes difficult to obtain, methods to sterilise them have been investigated. Gamma radiation, ethylene oxide or microwaves have been used. Viable organisms are successfully killed but spore-forming bacteria especially may remain intact when radiation doses are used that do not adversely affect the active substances.
The microbiological quality requirements of water are described in Sect. 23.3.1.
23.1.7.2 Bacterial Endotoxins and Pyrogens
Microbiological contamination of raw materials can lead to a finished product that does not meet the requirements for microbiological purity and in addition a high endotoxin level can be found leading to a pyrogenic response after intravenous injection.
The terms pyrogens and endotoxins (bacterial) are often used interchangeably. However, the Ph. Eur. makes a clear distinction between them:
Pyrogens are substances that cause a febrile reaction.
Endotoxins are lipopolysaccharides of the cell wall of gram-negative bacteria that causes fever when injected intravenously.
So not all pyrogens are endotoxins, see Sect. 19.3.4. Endotoxins are however the most common cause of toxic reactions that are attributed to contamination of pharmaceutical products with pyrogens; their pyrogenic activity is much greater than that of most other pyrogenic substances.
For approximately 80 raw materials the Ph. Eur. has set limits for endotoxin levels if they are intended for parenteral dosage forms. These include among others: dextran, sorbitol and mannitol, sodium chloride, trometamol, water for injections.
23.1.7.3 Prions
Raw materials especially of animal origin, or produced using reagents of animal origin, can be infected with prions, some of which are the cause of transmissible spongiform encephalitis (TSE). See Sect. 19.3.1 for information about the nature and pathogenicity of prions. Examples of raw materials that may be infected with prions are gelatin and fatty acids and fats of animal origin: oleic acid, magnesium stearate, glycerol, sorbitane esters, polysorbates and wool fat.
The deactivation of prions, if it can be achieved, requires aggressive interventions on the material, see also Sect. 19.3.1. Therefore it is essential to obtain the raw materials from a supplier that delivers a certificate, certifying that the raw material is free from TSE risk material. One can consult the EDQM site by the material name (through the field Databases and the Certification button) where manufacturers can display a TSE certificate for the requested substance [14].
23.1.8 Physico-chemical and Functionality-Related Characteristics
The Ph. Eur. monograph Pharmaceutical Preparations states: “When physico-chemical characteristics of active substances and functionality-related characteristics (FRCs) of excipients (e.g. particle-size distribution, viscosity, polymorphism) are critical in relation to their role in the manufacturing process and quality attributes of the pharmaceutical preparation, they must be identified and controlled.”
Ph. Eur. contains a monograph on FRCs. The chapter is not mandatory, neither is the FRC-section in specific monographs. But that does not mean that FRCs are unimportant. Especially in pharmaceutical development FRCs determine the design space of a medicinal product. Many excipients are manufactured by industries other than the pharmaceutical industry, so in many cases the excipient manufacturer has little knowledge about the pharmaceutical use of an excipient. Several methods to determine FRCs are described in Ph. Eur. e.g. Particle-size determination, Specific surface area by gas adsorption, Powder flow, Bulk density and tapped density, Wettability of porous solids including powders.
Two FRCs will be dealt in more detail: particle size and viscosity.
23.1.8.1 Particle Size
Particle size is an important physical quality property of raw materials. With regards to particles in this section a distinction is made between firstly loose crystals (primary particles) and secondly agglomerates: coagulated small particles (secondary particles) that have such a strong cohesion that they resist normal dispersion techniques and therefore are not easy to disperse in the production process.
The particle size determines to a large extent the dissolution rate and later in the use of the final product may determine the bioavailability of poorly soluble compounds. The stability of suspensions and the homogeneity of powder mixtures may also be influenced. For a detailed description of the importance and reduction of particle size we refer to Sect. 29.2. If a substance does not have the required degree of fineness for the intended process then it will be necessary to bring it to that degree.
The Ph. Eur. sets requirements for the particle size of raw materials in eye ointments and suspensions for injection. When the particle size is important for a raw material, it is common to display it in micrometres between parentheses after the name of the substance. The Ph. Eur. states in the monograph Sieve test that the fineness of a powder is expressed by the two sieve numbers, where at least 95 % of the powder passes the higher sieve size and not more than 40 % the lower sieve size (see Table 23.3), unless otherwise prescribed in the monograph. In the case of a single number, the Ph. Eur. means that at least 97 % of the powder passes the sieve size.
Table 23.3
Terminology concerning the degree of fineness of raw materials
Ph. Eur.: | ≥95 % m/m smaller than | ≤40 % m/m smaller than |
|---|---|---|
Coarse | 1,400 μm | 355 μm |
Moderately fine | 355 μm | 180 μm |
Fine | 180 μm | 125 μm |
Very fine | 125 μm | 90 μm |
In BP [15] additionally: | ||
|---|---|---|
Moderately coarse | 710 μm | 250 μm |
Microfine | ≥90 % m/m smaller than 45 μm | |
Superfine | ≥90 % m/m smaller than 10 μm | |
The monograph Sieves of the Ph. Eur. has a table with sizes and tolerances of a series of 18 sieves. The smallest sieve has (square) holes with sides of 38 μm, the largest holes with sides of 11.2 mm. The result of a sieve analysis is indicated as the percentage by weight of the substance that passes the sieve. Many raw materials are naturally fine (talc, starch) or will be delivered in the required degree of fineness, such as paracetamol (45) and salicylic acid (90).
Highly active substances are in most cases available sufficiently fine. The designation ‘micronised’ is not standardised, but a very good description is given by Møller [16]: 90 % of the number of particles <5 μm and all <25 μm. In this area of fineness, the given ‘particle size’ depends strongly on the method of measurement.
The particle size measurement of the raw materials is usually done by the supplier or the manufacturer. For the interpretation of those results it is necessary to know the principles of some methods. In the design phase, during in-process controls and at final testing, it is important to choose the particle size measurement method that is relevant to the property for which the particle size is investigated. Next to the determination of the particle size, a description of the nature of particles (crystals, agglomerates or aggregates) may be recommended.
As with all methods caution must be exercised to take a representative sample, both in terms of sampling location and size, for both the raw material and the product. A good overview of the different sampling methods is available in the literature [16].
With microscopy both the form (crystal, agglomerate) and the dimensions of a particle can be determined. The microscope must be equipped with ocular micrometers. With a normal light microscope dimensions can be determined for crystals or agglomerates of 5 μm to a few 100 μm.
It is possible but not easy to describe semi-quantitatively the particle size of a raw material by a microscopic method [16]. But generally other methods are used for the characterisation of powder fineness like sieving, air permeability or gas adsorption.
Sieving gives a visual check of the size by what passes through the sieve holes, in dry form or suspended in a liquid (wet sieving), under the influence of gravity or possibly using vacuum. The sieving result and its reproducibility not only depends on the particle size but also on the crystalline form, the degree and strength of agglomeration, the shape of the sieve openings, the flow properties, the sieve technique, the duration of the sieving process and the sample size.
In addition to the afore mentioned methods there are still special instrumental techniques applied such as the laser diffraction method for the determination of the particle size and the gas permeability method for the determination of the specific surface of a powder [17]. A further discussion of these methods is beyond the scope of this book. More information is available in textbooks [18].
Some substances are available in several degrees of fineness. The raw material triamcinolone acetonide is available as a micronised powder, to be used if triamcinolone acetonide is dispersed in a base. The micronisation may increase dissolution rate and hence bioavailability. It has, however, a strong agglomeration tendency and therefore cannot be processed as such. The 1 to 10 dispersion with rice starch contains micronised triamcinolone acetonide, whose particles don’t agglomerate any more due to the presence of rice starch and it may be preferred for the preparation of creams. Triamcinolone acetonide (crystalline form) as such can be used if it is dissolved in propylene glycol (for ointments) or in ethanol (for ear drops).
To improve the absorption and bioavailability, paracetamol in suppositories is processed as particles < 45 μm. In powders because of the greater density and better flow properties paracetamol (500–90) is used and for solutions either product can be used.
Microcrystalline tetracycline hydrochloride in micronised or microcrystalline form is applied in ointments, creams and eye ointments, while the coarser (‘heavy’) tetracycline hydrochloride is applied in capsules because of the greater density and in suspensions because of the greater chemical stability.
Microcrystalline cellulose (Avicel®, Pharmacel®) also exists as many variations of particle size. See Sect. 23.4.1.
Lactose has many variations in terms of particle size as well, each with their specific application in the food industry. The products for pharmaceutical use reflect only a small portion of the total volume of lactose produced by multinational companies worldwide. These multinationals indicate the particle size by using mesh seizes, as is usual in the food industry. The mesh size gives the number of openings per linear inch on a screen, see Table 23.4. Suppliers of pharmaceutical grade lactose usually specify their products based on sieve analyses, so expressing the result in μm.
Table 23.4
Conversion table suitable for some commonly used lactose types
Mesh size | Approximate size of opening |
|---|---|
80 | 177 μm |
100 | 149 μm |
200 | 74 μm |
325 | 44 μm |
400 | 37 μm |
450 | 32 μm |
23.1.8.2 Viscosity
Chemical quality indications are for instance important for cellulose derivatives. The quality depends on the length of the polymer chain and is usually indicated by the viscosity that is achieved in a 2 % solution. An example is hypromellose 4000 mPa.s. This quality is a highly viscous form. A 2 % solution of medium viscous forms of the cellulose derivatives usually have a viscosity of some hundreds and the low viscous quality of 10–100 mPa.s.
23.1.9 Mix-Up of Names
If names of substances are similar they can easily lead to mistakes. Well-known examples are: promazine and promethazine, salicylic acid and acetylsalicylic acid, sorbic acid and ascorbic acid, xylocain and xylometazolin, tetracycline and tetracain, Aerosil® and Avicel®, Oleum soya emulgatum and Oleum soya raffinatum etc.
A notorious source of confusion and ambiguity are the phosphates and their different ways of naming. Mistaking one for another in buffer solutions will lead to unexpected pHs. Mistaking the calcium phosphates may lead to wrong strengths in preparations. A short overview of the structural formula, name in Ph. Eur. and much used other names is given in Table 23.5.
Table 23.5
Overview of phosphates used in pharmaceutical practice
Structural formula | Name in Ph. Eur | Synonyms |
|---|---|---|
CaHPO4 | Calcium hydrogen phosphate anhydrous | Calcium phosphate, dibasic anhydrous |
CaHPO4.2H2O | Calcium hydrogen phosphate dihydrate | Calcium phosphate, dibasic dihydrate |
Ca3(PO4)2 | Calcium phosphate | Calcium phosphate, tribasic |
Calcium orthophosphate | ||
KH2PO4 | Potassium dihydrogen phosphate | Potassium phosphate, monobasic |
K2HPO4 | Dipotassium phosphate | Dibasic potassium phosphate |
NaH2PO4.2H2O | Sodium dihydrogen phosphate dihydrate | Sodium phosphate, monobasic |
Na2HPO4 | Disodium phosphate anhydrous | Disodium hydrogen phosphate |
Na2HPO4.2H2O | Disodium phosphate dihydrate | Sodium phosphate, dibasic (anhydrous, dihydrate, dodecahydrate) |
Na2HPO4.12H2O | Disodium phosphate dodecahydrate | |
Na3PO4.xH2O | – | Tribasic sodium phosphate |
New names in the Ph. Eur. also provide for confusion such as the two known forms of Cera lanette N and SX. Cera lanette N is now called Cetostearyl alcohol (type A) emulsifying (Alcohol cetylicus et stearilicus emulsificans A). Cera lanette SX is now called Cetostearyl alcohol (type B) emulsifying (Alcohol cetylicus et stearilicus emulsificans B). The mix-up is easily made with Cetostearylalcohol (Alcohol cetylicus et stearilicus, a mixture of equal parts cetyl-and stearyl alcohol).
Ph. Eur. will change names from time to time. Sometimes it is quite obvious and there is no ambiguity between the old and the new name, just a shift in the alphabetical order (examples are sodium diclofenac which has become diclofenac sodium, ethyl parahydroxybenzoate sodium that has become sodium ethyl parahydroxybenzoate). But sometimes, especially with hydrates (ferrous sulfate is now ferrous sulfate heptahydrate), the substance is a different one and sometimes the name will change completely but the substance remains the same e.g. Glycerol triacetate becomes Triacetin, Chloramine becomes Tosylchloramine sodium.
Differences in numbers and figures in the name can cause confusion. Cetostearyl alcohol (type A) differs from Cetostearyl alcohol (type B) concerning the emulsifier (type A: sodium cetostearyl sulfate; type B: sodium lauryl sulfate). The macrogols are followed by a number, which indicates the average molecular mass of the chain.
Vitamin A (Retinol) and Vitamin A acid (Tretinoin) are different substances and cannot be used instead of each other because they have a different pharmacotherapeutic application. Folic acid (Acidum folicum) and Folinic acid (Acidum folinicum) are closely related substances but differ significantly in effective strength. Tocopherol esters are applied as vitamin E; tocopherol itself is especially applied as antioxidant.
It is necessary to be alert to the existence of different esters or ethers of a substance and different amounts of water of crystallisation. The availability of two or three different forms of a raw material can easily lead to mix-ups. For examples refer to Table 23.1 and to Sect. 23.1.5.
Sometimes the name of a raw material is incomplete and therefore not unambiguous. Polymyxin can be polymyxin B or polymyxin E (colistin). If the designation vitamin D is used, it may be assumed that cholecalciferol, vitamin D3, is meant and not the less effective ergocalciferol, vitamin D2.
23.2 Quality, Stability and Shelf Life
Some raw materials degrade or otherwise lose quality. This may be the result of:
Hygroscopicity; for example calcium chloride, docusate sodium
Efflorescence, which is losing crystal water; it occurs especially with sulfates and in particular with zinc sulfate, and with substances with a high crystal water content such as disodium phosphate dodecahydrate
Oxidation: for example cholecalciferol, ferro compounds, tretinoin, and catecholamines, fatty oils
Microbiological spoilage: for example water, starch, paracetamol, amphotericin
The purity of the raw material will affect the active substance content of the finished product and requires factorisation. This means: correcting the quantity of a raw material to be weighed for the active substance content deviating from 100 % in the raw material. The content requirements of the finished product and the recommendations concerning factorisation can be found in Sect. 32.4.
The pack sizes of raw materials are often such that the contents will not be used in its entirety, but in parts. Along with the substance’s properties, its packing, storage and use determine the shelf life of the raw material in the pharmacy. The shelf life of a raw material is displayed on the package by an expiration date. This applies to the shelf life at the standardised storage conditions for this raw material, in an unopened packaging. If these storage conditions are not maintained in practice, or when the packaging is opened frequently, it is likely that the label claim is no longer applicable for the content. This phenomenon occurs especially in efflorescing or strongly hygroscopic substances and also substances sensitive for oxidation such as fatty oils with many unsaturated fatty acids.
Prednisolone sodium phosphate and dexamethasone sodium phosphate are examples. They always contain a quantity of water and ethanol which is factorised in the standardised worksheets. Both substances can attract more moisture than specified according to the label. In addition, the attracted moisture accelerates the decomposition. For small scale preparation raw materials suppliers sell these substances in small quantities in a well closed container, sometimes filled under a nitrogen atmosphere, in order to remain compliant to the specifications during the listed shelf life. Within a few weeks after opening the container in the pharmacy the substance will no longer meet the specifications as a result of degradation, unless the substance is stored in a desiccator in the fridge. It is therefore recommended to process the whole pack into the final product at one time.
The water content of zinc sulfate.7H2O can decrease by efflorescence. The water may condense at the top of the container and to the inside of the lid. This water can be reabsorbed by shaking well.
Both forms of calcium chloride (CaCl2.6H2O and CaCl2.2H2O) can attract water by hygroscopicity and subsequently dissolve in the attracted water.
Water is able to evaporate from chlorhexidine digluconate solution 20 %. This may lead to a high content in chlorhexidine creams and mouthwashes. An use-by period of 1 year is therefore recommended for this raw material.
Caution must be exercised with these kinds of substances and they need to be stored properly and not being used if the expiry date is exceeded. The influence of light should not be underestimated. The Ph. Eur. states for many substances, for example tretinoin, phytomenadione, corticosteroids and benzodiazepines that they must be protected against light. In oils and fats oxidation and peroxide formation is increased by light. Sorbic acid in solution (so also in preparations) is sensitive to light.
Proteins is a group of active substances that will be used increasingly in the future. Proteins are large vulnerable molecules with a complex structure. Their many functional groups render proteins very sensitive to chemical and physical degradation. These can lead to disintegration of the molecule or may cause a change in the tertiary structure. In both cases, the biological activity is lost, see Sect. 18.4.1. The chemical processes that can play a part are: oxidation, hydrolysis, desamidation, disulfide conversion, beta-elimination, racemisation. Physical processes that may occur are: precipitation, adsorption, absorption, aggregation and denaturation. This means, that many factors can influence the stability of proteins. Acidity, oxygen and light are known factors or catalysts of degradation reactions. Temperature plays a part in many of degradation processes. Freeze-thaw cycles may greatly affect the material. In addition, the type of degradation reaction will depend on the temperature.
23.3 Solvents
23.3.1 Water
Various water qualities that are used in the preparation or reconstitution of medicinal products are described in the Ph. Eur. and in an EMA Note for Guidance [19]. See for the overview Table 23.6. The production of water for pharmaceutical purposes is described in Sect. 28.3 and storage and distribution in Sect. 27.5.2.
Table 23.6
Constituents and impurities which, as a result of the preparation method and storage conditions, may be found in the different types of water
Type of water | Subcategory | Minerals | Heavy metals | Gases | Micro-organisms | Organic substances* |
|---|---|---|---|---|---|---|
Potable water (tap water) | + | + | + | + | + | |
Water, purified Ph. Eur. (Aqua purificata) in two forms: | Demineralised water | – | +(−) | + | ++ | + |
Distilled water | – | – | + | + | + | |
Purified water in bulk | ||||||
Reverse-osmosis water (RO-water) | – | – | + | + | + | |
Purified water in containers | ||||||
Water, highly purified Ph. Eur. (Aqua valde purificata) | – | – | + | –(+) | – | |
Water for injections Ph. Eur. (Aqua ad iniectabile) (WFI) | Water for injections in bulk | – | – | – | –(+) | – |
Sterilised water for injections | – | – | – | – | – |
23.3.1.1 Potable Water
Potable water (Tap water, Aqua, Aqua communis, Water for human consumption) is as such a raw material for purified water in small-scale preparation and it may be used for the early stages of cleaning pharmaceutical equipment. It has the advantage that, if there is a good flow rate through the pipes, in most European countries it usually has a reasonable microbiological quality.
Potable water contains as additives sodium, calcium, magnesium, chloride and carbonate ions and a number of other ions in very low concentrations. The bivalent ions of calcium and magnesium are the cause of hardness in water. The hardness of water is specified as hardness degrees, see Table 23.7.
Table 23.7
Water hardness and corresponding calcium salt concentration
mg Ca/L | mg CaCO3/L | English degrees | French degrees | German degrees | Hardness |
|---|---|---|---|---|---|
<30 | <75 | <5.3 | <7.5 | <4.2 | Very soft |
30–50 | 75–125 | 5.3–8.8 | 7.5–12.5 | 4.2–7.0 | Soft |
50–100 | 125–250 | 8.8–17.5 | 12.5–25.0 | 7.0–14.0 | Moderately hard |
100–150 | 250–375 | 17.5–26.3 | 25.0–37.5 | 14.0–21.0 | Hard |
>150 | >375 | >26.3 | >37.5 | >21.0 | Very hard |
Hard water will form precipitations of calcium and magnesium salts of the anions that are dissolved in the water. The hardness can be subject to regional differences. The water company should be consulted (or the internet searched) for the hardness of the water it is providing.
Among the trace ions, the heavy metals ions are of particular pharmaceutical interest. For instance copper and lead can occur in potable water, since pipes can be made of these materials. To limit the electrolytic formation of these ions, the pipes are now often made of plastic (PVC) and the earth wire is no longer connected to the water pipe.
The common air gases, carbon dioxide, oxygen, and nitrogen, are also dissolved in water. The dissolved oxygen may decrease the stability of oxidisable active substances. Carbonic acid will form poorly soluble carbonates with many bivalent positive ions.
Fresh draught potable water meets the following microbiological quality requirements, provided that it flows well through the pipes and that there is no holding tank: not more than 100 micro-organisms per mL, and Escherichia coli <1 per 100 mL. In practice the contamination of fresh potable water will not exceed 10 CFU/mL (CFU = colony forming units1). This means that fresh draught potable water in microbiological terms is suitable for the manufacture of non-sterile medicinal products; the Ph. Eur. limit for these products being 100 CFU/mL. Do not use hot water from the tap, because it might come from a holding tank; prepare it by cold potable water and heat if necessary.
It is necessary to check occasionally the quality of potable water. Especially in hospital pharmacies one should anticipate the presence of still water, due to the presence of holding tanks. In those circumstances, germ growth will occur such that the limit value of 100 CFU/mL may be exceeded. Pharmacy should have access to water supplied directly from the mains supply. Nevertheless the quality of the water supplied by the water company is not under the control of the pharmacy.
Potable water can be used for the various antibiotic oral mixtures that are reconstituted from the dry granulate.
23.3.1.2 Purified Water
Purified water is water that has been purified from the hardness minerals by one of the methods mentioned by Ph. Eur. Ph. Eur. has two types of Purified Water: Purified Water in bulk and Purified Water in containers. It is used in the pharmacy in several ways: as a raw material for the manufacture of non-sterile medicines, sterile medicines that are not necessarily free of endotoxins, in water baths, as cooling water for steam sterilisers, and sometimes for rinsing glassware or packaging material. For Highly purified water Ph. Eur. the microbiological and endotoxin requirements are the same as for Water for injections.
Chemical Purity
The monograph Water, purified of the Ph. Eur. set limits for total organic carbon (TOC) or oxidisable substances, endotoxins (if intended for dialysis solutions), conductivity and the following ions: nitrates, aluminium (if intended for dialysis solutions) and heavy metals. In addition it is necessary to perform microbiological monitoring during production and storage. For purified water in containers, the Ph.Eur. adds limit tests for acidity or alkalinity, chlorides, sulfates, oxidisable substances (mandatory, no choice of TOC), ammonium, calcium and magnesium, residue on evaporation and microbiological contamination. The Ph. Eur. also requires that the conductivity is measured. At 20 °C the conductivity may not exceed the value of 4.3 microS.cm−1.The measurement may be in-line or off-line. In-line measurement (see Sect. 27.5.2) enables a quick and permanent monitoring of the removal of sufficient minerals. If the water complies with the requirement for the conductivity for Water for Injections (1.1 microS.cm−1) the test for heavy metals does not need be carried out.
Measuring the pH with a pH meter is only possible if the conductivity is increased by the addition of a little potassium chloride. Distilled water has a low pH, which quite often is caused by dissolved carbon dioxide. This is removed during the distillation process but it can re-enter by diffusion. This is a strong acid with a limited solubility. It is partly the cause of distilled water’s aggressiveness towards base metals (beware with aluminium). Dissolved gases may also be found in demineralised water since the process is not designed to remove them. neither is the demineralisation process designed to remove heavy metals, so also these can be found.
Carbon dioxide dissolves mainly in the form of the carbon dioxide monohydrate (CO2.H2O) which is in equilibrium with carbonic acid (H2CO3). The balance, however, lies predominantly to the hydrate side. Insofar as carbonic acid is formed, this is fully split into ions and thus a strong acid. However, it usually behaves as a weak acid because of the natural shift to the hydrate side.
Microbiological Purity
In the monograph for Water, purified a specification for the microbiological quality is included: not more than 100 CFU/mL (TAMC, see Sect. 19.6). This corresponds to the strictest requirement for the microbiological contamination of preparations. In general the requirements for raw materials are stricter than those for the preparations, but not in this case. The absence of E. coli is essential, but the starting material (potable water) has been checked already and the growth of these organisms in the systems of water purification is unlikely. For measuring the microbiological quality of water the method (the nutrient medium and the temperature), should be chosen, see Sect. 19.6. The requirement of microbiological purity is to be met at start of production by distillation but if this water is prepared by demineralisation or is kept longer than 24 h after production, then a microbiological reducing treatment by heating or filtration is necessary.
A test on bacterial endotoxins is also included in the Ph. Eur. because of water for the production of dialysis preparations.
For applications requiring water of high microbiological quality the Ph. Eur. includes the monograph Water, highly purified. The ‘requirement’ (the pharmacopoeia states an action limit to production) is 10 CFU/100 mL. This requirement corresponds to that of Water for injections; yet Highly Purified Water is considered unacceptable for use as Water for Injections. It can be used when medicinal products administered by nebulisation are required to be sterile and non-pyrogenic and for the final rinse of equipment, containers and closures for sterile products [19]. The Pharmacopoeia lists it as one of the types of water that can be used in gene transfer.
23.3.1.3 Water for Injections
Water for injections is prepared according to the Ph. Eur. by distillation of potable or purified water from a device of neutral glass, quartz or a suitable metal. The device must be fitted with an anti-splashing device to prevent contamination of distilled water with non-distilled water.
Water for injections in bulk must comply with the requirements as formulated for Purified water. It must also be produced free of bacterial endotoxins and stored such that it remains free of them. It can be used for parenteral products and irrigations that are terminally sterilised. Water for injections, sterilised, must meet the requirements of sterility (sterility test) and the bacterial endotoxin content must not exceed 0.25 IU per mL. This is used for parenteral products and irrigations that are not terminally sterilised and for dissolving powders for injection immediately before use.
Purification should therefore only take place in this case by distillation because demineralisation and reverse osmosis, as purification processes, are considered too risky from a microbiological point of view and because of the risk of developing bacterial endotoxins. However, in the Japanese Pharmacopoeia reverse osmosis is now an accepted method and in Europe there are ongoing discussions to accept reverse osmosis after all [20].
When collecting and storing water, the growth of micro-organisms and the increase of endotoxins as bacterial waste products must be prevented. That means it is either collected and kept at a temperature higher than approximately 70 °C until it is processed (Water for injections in bulk), or it is sterilised, either directly or after filling into vials or ampoules (Sterilised water for injections, Aqua ad iniectabile sterilisata). See further Sect. 27.5.2.
Purified water can also be purchased. In that case Sterile purified water or Sterilised water for injections is recommended because of the insecure shelf life and usage period of non-sterile water. Table 23.8 provides practical, not official, guidelines for keeping water in small-scale situations without a storage and distribution installation.
Table 23.8
Guidelines for keeping water for non-sterile medicines
Method of preparation + way of storage: | Storage temperature: | Storage period: |
|---|---|---|
Distillation | ||
Storage in closed bottle | 2–8 °C | 2 weeks |
Bottle after opening | 15–25 °C | 24 h |
Vessel from which it is tapped | 15–25 °C | 24 h |
Demineralisation plus boiling | 15–25 °C | 24 h |
Demineralisation with membrane filtration | 15–25 °C | None: use immediately |
Sterile purified water | ||
Closed bottle | 15–25 °C | 3 years |
Bottle after opening | 15–25 °C | 24 h |
Sterilised water for injections | ||
Closed bottle, sterilised | 2–30 °C | 3 years |
Bottle after opening | 2–30 °C | 24 h |
A special quality of water is described in addition in the Ph. Eur.: Water for the dilution of concentrated haemodialysis preparations. This monograph is included for information purposes and does not have the same legal force as the other pharmacopoeial monographs. Nephrology associations may have elaborated supplementary guidelines, such as in the UK [21] and ISO13959:2009. See also Sect. 14.5.
Special technical demands are applied to water which is used as cooling water for sterilisation equipment and other devices used in health care.
23.3.2 Ethanol
In pharmacy ethanol is usually used as a solvent and sometimes as a preservative. It can also be used as a disinfectant, mainly in a concentration of 70 % V/V in water. The excess use of alcohol as a cleaning agent has been reduced after it was registered as being mutagenic on inhalation. Although the Occupational Exposure Limit (OEL) of ethanol is rather high in many European countries: about 1,000 mg/m3 as Time Weighted Average (TWA)-8 h and 1,900 mg/m3 as TWA-15 min, this level can be easily exceeded in a small room, see Sect. 26.7.2.
The concentration of ethanol in water is still given in volume/volume percentage. When adding a mL ethanol to b mL of water the final volume of the solution is smaller than the sum of a + b. This phenomenon is called contraction. The alcoholimetric tables of the Ph. Eur. provide sufficient information to prepare a variety of concentrations. Next to the described concentrations in Ph. Eur. (absolute and 96 %), ethanol 70 % V/V (62.4 % m/m) is used for preparation purposes.
Ethanol as such is not automatically sterile. 50–70 % V/V ethanol/water is maximally bactericidal. Higher concentrations can cause some micro-organisms to transform into spores, which are able to survive. If the ethanol concentration of a liquid is higher than 15 % (V/V) in combination with a pH lower than 7, or if the concentration is higher than 18 % (V/V) in combination with a pH higher than 7, no preservative needs to be added. These conditions make the liquid self-preserving. Alcohol 96 % V/V can be sterilised by distillation. Filtration by ethanol-resistant membrane filters with pore size 0.2 μm will reduce the number of micro-organisms considerably. Mixtures with sufficient water content can be sterilised by autoclaving. Hydrogen peroxide may be added as a sporicide.
23.3.2.1 Denaturated Ethanol
In most countries excise duty must be paid on pure ethanol. Therefore in some countries alcohol that is intended for external application or disinfection purposes (including disinfection of skin) is usually denaturated: rendered unsuitable for consumption. When denaturated, the duty can (partly) be exempted. The exemption of duty may be bound to licenses stating the amount of ethanol that is bought through the year and the agents used to denaturate.
In Netherlands a usual way of denaturating alcohol is the addition of 5 mL of methylethylketone, 25 mg denatonium benzoate and 2.5 mL of synthetic bergamot oil to 1 L alcohol 96 % V/V. The methylethylketone renders the solution azeotropic thereby making re-distillation of pure ethanol impossible. Denatonium benzoate has an intensely bitter taste, thus creating an unpalatable drink. A similar method of denaturating ethanol with methylethylketone and sometimes addition of a dye occurs in Switzerland, Poland, Norway, and Czech Republic. In Kosovo and Turkey denaturating is not common nor is it carried out in Croatia, where ethanol used in pharmacies is free of tax.
After denaturation the alcohol is of course unfit for use in oral preparations.
The alcohol denatured with methylethylketone cannot be used as a solvent for iodine spirit because methylethylketone, like acetone, will complex with iodine. This complex irritates the eyes of people using it or applying it on the skin.
The addition of 5 % methanol may be considered by authorities to be sufficient to denaturate ethanol. Ethanol denaturated with 5 % methanol is commonly used in cosmetics despite the fact that methanol (and also methylethylketone) is, according to the H-statements (see Sect. 26.3.2) considered to be too toxic when regularly applied to the skin of workers.
23.3.3 Glycols and Glycerol
Glycols are diols (alcohols with two hydroxyl groups). Ethylene glycol has no pharmaceutical application. It is toxic because it strongly binds calcium.
Propylene glycol is a diol (propane-1, 2-diol). It is in use in cutaneous preparations, dissolved in the aqueous phase, as a humectant, to slow down a dehydration process. Other applications are the use as softening agent and as vehicle for ear drops intended for the external auditory meatus (see Sect. 9.5.3).
In mixtures with ethanol and water it is a solvent for active substances with low water solubility such as digoxin, diazepam, barbiturates and phenytoin (see also Sect. 18.1.3). The ratio water – ethanol – propylene glycol (or glycerol) depends on the substance to be dissolved. Propylene glycol concentrations higher than approximately 15 % are self-preserving. Depending on the formulation of the preparation, concentrations from 13 % may be sufficient for preservation. Mixtures with water can be sterilised by steam sterilisation. Propylene glycol is toxic in (chronic) oral use by children and patients with poor renal function [22].
In 1937 the Elixir Sulfanilamide disaster occurred, one of the most consequential mass poisonings of the twentieth century. This tragedy occurred when propylene glycol was used as the diluent in the formulation of Elixir Sulfanilamide. More than 100 patients died. At that moment premarketing toxicity testing was not required. In reaction to this calamity, the U.S. Congress passed the 1938 Federal Food, Drug and Cosmetic Act, which required proof of safety before the release of a new drug [23]. The FDA was established.
Glycerol is a triol (propane-1,2,3-triol) and very hygroscopic. Glycerol 85 % is less hygroscopic and thus less perishable. The applications are similar to those of propylene glycol. When used in oral liquid preparations the taste is slightly sweet and better than that of propylene glycol. Glycerol is more hydrophilic than propylene glycol; self-preserving concentration is higher than approximately 30 %, so significantly higher than that of propylene glycol. Glycerol/water mixtures are sterilised by steam sterilisation.
23.3.4 Macrogols
Macrogol (polyethylene glycol, PEG) is a polymer of ethylene oxide, see Fig. 23.1.
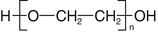
Fig. 23.1
The formula of macrogol
The chain length and the molecular mass depend on the polymerisation degree n. Macrogol 400 has a molecular mass of 400 in a polymerisation degree n = 8. Below a molecular mass of 700 the macrogols are liquid, above 1000 they are solid. Macrogols are used as a basis for hydrophilic ointments (Sect. 12.7.8) and as water-soluble suppository bases (Sect. 11.4.5). During storage macrogols can be slightly oxidised by oxygen from the air. For that reason they are incompatible with oxidisable active substances, especially if those are applied in low concentrations (e.g. ergotamine).
Macrogols with a low degree of polymerisation(<2000) are hygroscopic [5].
23.3.5 Fatty Oils, Fat, Waxes and Paraffin Waxes
Oils and fats are triglycerides (esters of glycerol with three fatty acids, mainly palmitic acid, stearic acid and oleic acid). The melting point and consistency decrease with decreasing degree of saturation and increase with larger chain length. The melting point of a saturated substance is higher than that of the corresponding unsaturated substance.
Esterification of glycerol with saturated short chain fatty acids and unsaturated short-and medium chain fatty acids usually results in (at room temperature) liquid oils; longer fatty acids give hard fats. Table 23.9 shows several examples of natural and synthetic oils and fats.
Table 23.9
Some commonly used fats and oils
Vegetable, unsaturated, liquid | Almond oil |
Arachis oil | |
Olive oil | |
Rapeseed oil | |
Sesame oil | |
Soya-bean oil | |
Vegetable, unsaturated, solid | Cocoa butter |
Animal, saturated,a solid | Lard |
Synthetic, saturated, liquid | Triglycerides, medium chain (Miglyol 812®) |
Synthetic, saturated, solid | Hard fat |
Rapeseed oil and sesame oil, given their sensitising properties, are no longer applied in cutaneous preparations. They can cause contact allergy. Groundnut oil is made from peanuts and may cause, when in non-purified form, peanut-allergy. When using the purified, pharmaceutical form, allergic reactions will not occur [24].
Hard Fat consists of a mixture of mono-, di- and triglycerides. It is used as a lipid suppository base. More information can be found in Sect. 11.4.4.
Ph. Eur. contains assays, for acid, hydroxyl, iodine and peroxide values, to determine the qualities of oils and fats.
The acid value is a measure of the amount of free fatty acids in the fat. An appropriate acid number, so a correct amount of free fatty acids, of groundnut oil is important in the preparation of Zinc oxide calcium hydroxide weak paste FNA (see Table 12.39). The ideal is 0.25–0.5. If the acid value is low (e.g. 0.06, for a just opened package) then free oleic acid must be added. At too high an acid number the emulsion breaks.
The hydroxyl value is a measure of the number of acylable places and thus the content of mono-and diglycerides. A suppository base (Hard fat) with a low hydroxyl value (<15) is less damaging to the stability of acetylsalicylic acid and ergotamine tartrate than bases with a higher hydroxyl value. Witepsol H15 has a sufficiently low hydroxyl value (5–15) for this preparation. Bases with a high hydroxyl value (up to 50) have a better emulsifying ability and can include aqueous systems in the form of a water-in-oil-emulsion.
The iodine value is a measure of the number of double bonds. For Hard fat the Ph. Eur. requires a value ≤ 3.0.
The peroxide value is a measure of the number of peroxide bridges in the fat. Fats or oils with a low peroxide number (≤0.5) are used for the processing of easily oxidisable substances. Examples are ergotamine tartrate and chlorpromazine hydrochloride in fatty suppositories.
The saponification value is a measure of the number of available saponifiable ester bonds. Also the unsaponifiable matter may still be used as a property.
For each fatty oil or fat the requirements are specified in the relevant monograph.
Unsaturated fats (high iodine value: >4) are sensitive to oxidation by peroxides; becoming rancid. Adding antioxidants (radical scavengers, examples: butylhydroxyanisole and butyl hydroxytoluene, see Sect. 23.9) will prevent this.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


