A. Stock preparations
Categories
A1 Sterile
Aseptic preparations (vials, ampoules, infusion fluids in bags, prefilled syringes)
III
Autoclavable preparations (vials, ampoules, infusion fluids in bags or bottles)
III
Eye drops
III or II
Sterile ointments and creams
III
A2 Non-sterile
Packaging e.g. in Unit Dose
III
Tabletting
III
Fluids (solutions and suspensions)
III
Ointments and creams
III
Suppositories
III
B. Extemporaneous preparations | |
|---|---|
B1 Sterile | |
Aseptic handling (complex, see Sect. 31.3.2) | II |
Total parenteral nutrition, medication cassettes | |
Aseptic handling (hazardous, see Sect. 31.3.5) | |
Antineoplastics, radiopharmaceuticals | |
Simple aseptic handling; short shelf-life | I |
B2 Non-sterile | |
Capsules, suppositories, fluids, ointments and creams | I |
Reconstitution of licensed pharmaceutical preparations | I |
27.2 Design
27.2.1 Main Layout Considerations
Starting from the classification of preparation processes a rough preliminary design is drafted indicating the position of production areas, their interrelationship and logistics. The relations between departments and premises, flow of goods and persons are plotted. This start document is preferably formulated by the one who will be responsible in future for the preparation processes.
The preliminary design should be drawn up based on the following considerations:
The extent of the preparation department: Available premises usually are confined. However, sufficient room must be available for apparatus and materials. Apparatus should be stored in a way that minimises contamination. Therefore, a separate accommodation near the preparation premises is preferred over the placement in the preparation room itself.
Sufficient room should be available for the operators to work efficiently and safely.
Avoiding crossing routes for personnel and goods: In a pharmacy several products are usually being prepared on the same day. This brings about a higher risk of cross contamination when compared to a large pharmaceutical industry producing only one product on a dedicated production line. Inadvertent mixing up of a sterile and a not yet sterilised product or of products of different batches constitutes a similar threat.
Entrance: The entrance to premises for preparation should be provided by means of, preferably sex-separated, gowning rooms for staff and a separate air locked entrance exclusively for goods. If necessary the separation of the sexes might be achieved by separating in time. However it should be realised that this may jeopardise the efficiency of the production.
A correct constructional design has to be derived from the process and will prevent any confluence or crossing of routes. However, in practice crossing cannot always be avoided. In that case procedural solutions should warrant that all goods are to be labelled and packed securely (e.g. in closed boxes) before being exposed to crossing lines. Routing questions at the design of a layout of premises will frequently be with regard to processes such as weighing raw materials, sampling, input and output to and from any temporarily deposit store for intermediates or quarantine, cleaning of reusable utensils and label printing.
For example, it is important to consider if the weighing process will or will not be carried out in a centralised weighing room thus requiring subsequent transport of the labelled portions to the preparation room. In a centralised weighing room, thorough precautions should be taken to prevent cross contamination of the raw materials. The same goes when two workers use the same set of weighing apparatus in a pharmacy.
The location of the preparation department in relation to logistic functions, etc. For example radiopharmaceuticals (see Sect. 15.6.1) should be prepared nearby or at the nuclear medicine department.
The required apparatus and utensils, the preparation processes they are used for and the required provisions such as air conditioning, clean water, electricity, compressed air and gasses. For extremely hazardous preparations (antineoplastics, radiopharmaceuticals, see Sect. 26.3.5) and for sterile preparations separate premises may be required to protect products and operators adequately, as is also laid down in GMP. Scaling up of the processes may lead to partitioning of the rooms into areas for dedicated functions.
After classifying the preparation processes and the corresponding premises and its outlines the preliminary design will be drafted. Some examples are given below for a few preparation processes.
27.2.2 Sterile Stock Preparations
Separate rooms are necessary for the preparation and for filling into e.g. infusion bags or ampoules prior to final heat sterilisation. In addition, one or more sterilisers with corresponding technical rooms (including the access) are needed.
The premises for the most critical preparation steps (e.g. filling) must be built as a clean room (see Sect. 27.3) and require a controllable air conditioning installation. Those premises must comply with GMP class C (see Sect. 27.4.2 and Table 27.2) [1].
Table 27.2
Grades of air conditioning according to GMP
Maximum permitted number of particles per m3 equal to or greater than the tabulated size | ||||
|---|---|---|---|---|
At rest | In operation | |||
Grade | 0.5 μm | 5.0 μm | 0.5 μm | 5.0 μm |
A | 3,520 | 20 | 3,520 | 20 |
B | 3,520 | 29 | 352,000 | 2,900 |
C | 352,000 | 2,900 | 3,520,000 | 29,000 |
D | 3,520,000 | 29,000 | Not defined | Not defined |
Sterile stock preparations usually require such high volumes of water that separate technical installation premises are required to accommodate the installation for the production of water for injections.
Finally, premises for examining and for packaging and labelling are necessary.
Appropriate rooms (laboratories) for pharmaceutical microbiological control and for chemical quality control should not be left out. However the possibility of microbiological contamination from this laboratory requires a completely separated air handling.
Alternatively the microbiological control could be out-sourced implying yet other, more procedural complications such as the need for Service Level Agreements and meeting the requirements of GMP Chap. 7 (Outsourced activities).
27.2.3 Aseptic Stock Preparations
In premises for aseptic preparation from sterile raw materials the preparation room usually is combined with the filling room because carrying the bulk product into another room will introduce an additional contamination risk. In this situation a separate room for preliminary operations, e.g. disinfection of utensils and surfaces of containers, is required.
At the most critical places, especially at the filling point of the aseptic fluids, the premises must meet GMP class A conditions [1]. A class A condition usually needs a background condition of class B to be able to maintain the class A condition during operation. However exceptions to this rule can be warranted, e.g. using an isolator.
Classified premises are expensive both in procurement and maintenance. Therefore, it is justified to analyse each preparation process for all critical steps and to limit their number as far as possible.
Premises for examining and for packaging and labelling can be combined with those for sterile stock preparations.
27.2.4 Aseptic Extemporaneous Preparations
Also in this situation a separate room is required for preliminary operations, e.g. disinfection of utensils and surfaces of materials. In the preparation room medicines are reconstituted, e.g. filling of syringes, infusion bags, medication cartridges, disposable infusion pumps and irrigations. In addition, parenteral nutrition fluids, antineoplastics, radiopharmaceuticals and eye preparations may be prepared in these premises. Radiopharmaceuticals and other very hazardous products, may require dedicated rooms and containment conditions (see Sect. 26.7).
For aseptic handling in closed systems a cabinet with unidirectional airflow (LAF or safety cabinet) or an isolator can be used (see Sect. 28.3). The requirements for the background room depend on national guidelines and on the types of containment in the cabinets. Adjustments are allowed but must be based on a risk assessment.
Strictly, air quality class A cannot be claimed if the background qualification is less than class B. However this class A safeguard is strictly only required for aseptic stock preparation and any aseptic handling that is executed with non-closed systems. Under specific conditions a class C condition as background might be acceptable. On the basis of a thorough risk assessment and a monitoring and validation program even a class D background for well-defined aseptic handling may be justified, see Sect. 31.3.4.
27.2.5 Non-sterile Stock Production
This department can be divided into separate premises for solid (dusty), semisolid and fluid preparations. Preparation processes with inflammable substances may require specific provisions such as a fume cupboard or more intensive ventilation. Premises for tableting and similar dust-producing preparation processes should be equipped with a suitable installation for air conditioning and dust exhaustion. Applying a negative pressure will prevent dust from active substances escaping from the preparation room. Even for repackaging activities (e.g. into unit dose package) a separate room is to be preferred. Specific GMP classification A-D is not required for non-sterile preparations. However the GMP principles such as cleanability are applicable. Therefore, in practice, facilities for non-sterile preparation are often classified as grade D.
27.2.6 Non-sterile Extemporaneous Preparations
The underlying principles for premises for non-sterile stock preparations should be used for extemporaneous preparations as well. Preparation activities in a community pharmacy usually are confined to reconstitution, aseptic handling, manipulation of licensed medical products and non-sterile preparation from raw materials. The avoidance of crossing process lines in small-scale situations is a challenge but it is almost impossible to prevent crossing lines just by the layout of the premises. Routing has to be specified by procedures and organisational measures have to be taken, e.g. working with trays or closed boxes per activity.
The requirements for the premises may turn out to be quite moderate, provided that they are based on a well-documented risk assessment. It will at least imply that premises shall be exclusively dedicated for preparation activities, e.g. shall not give direct access to toilets and will have to be physically separated from any public area. The layout should not compromise a logical sequence of activities. Specific gowning and cleaning procedures must apply.
Attention should be paid to the ventilation of any premise and its upkeep. In newly built premises, the ventilation capacity might have been minimised due to energy saving policy, especially in northern countries. Therefore the risk has to be assessed that the ventilation might fail to meet minimal requirements. Any so-called ‘natural ventilation’ might clear the way for insects to enter, unless specific measures are taken (e.g. insect screens). Air quality can be improved by using a recirculating dust exhaust cabinet which should be available in every community pharmacy, e.g. for the reconstitution of antibiotic oral liquids.
27.2.7 Storage Rooms
Apparatus to achieve specific, usually low temperature, storage conditions are described in Sect. 28.9. The design of storage rooms should take into account that the temperature should not exceed 25 °C for prolonged periods (see Sect. 36.9.4). This may involve the necessity of air conditioning. Additional provisions should be provided to avoid long lasting humidity levels over 60 % RV or below 20 % RV or penetration of direct sunlight.
Specific conditions, like lockable safety cupboards, may be required for the storage of any specific class of hazardous (e.g. inflammable or poisonous) products.
A properly considered location of storage rooms, separated quarantine storage and laboratory may ease routing very much.
There should be ample space for input and output of goods without the need of rearranging the goods at each occasion (first-in first-out principle). Especially the manoeuvring of pallets should be accounted for.
Empty packing material is usually wrapped in airtight wrappings or transport casings. However after opening the wrap, packing materials are open to the rather unconditioned environment of the storage room and thus open for pollution with dust or even insects. So either only complete units have to be used or the remaining materials should be rewrapped or repacked.
Storage rooms should be kept clean at a normal household level and vermin-free. No specific additional demands are made because the package of the products should protect them sufficiently against any contamination.
27.3 User Requirements Specification
As soon as the draft design, the program of requirements and a thorough (risk) analysis of the anticipated production activities is gathered, this information should be ‘translated’, usually by professional advisors, into a keynote document or user requirements specification (URS). This document should describe and specify in detail the required situation after the (re-)building. It should clearly underpin the listed demands and points of departure, which for their part should be traceable to relevant legislation. In addition the URS should, after realisation of the building, offer direct control points to all critical aspects of the intended processes. The conformity of the final situation to the original plan must be proven which only can be done by checking systematically all critical control points of the completed premises to the original plan as previously specified in the URS.
The delivery of this evidence is called the performance qualification (PQ), see further Sect. 34.15. It is important to pay attention to the future PQ as, in contrast to other qualifications, the PQ cannot be outsourced. A well-formulated URS takes account of the implementation of the future PQ tests and therefore its tenor extends far beyond a more trivial ‘Plan of Demands’.
The URS should account for regulatory demands of GMP, Occupational Safety and Health and Environmental Care. Additionally preparation demands, required apparatus, HVAC (installations for Heating, Ventilation and Air Conditioning) controls, routing of personal and goods as well as gowning and cleaning instructions should be specified. It should be stated explicitly which regulations (e.g. GMP, ISO standards, Occupational Safety and Health legislation) do apply, thus delimiting the legislative framework. By adding the prefix ‘c-‘ (current) to the name of any law (e.g. c-GMP) it is stated that the regulation may be developing during the life of the premises and that always the most recent version has to be consulted.
Clean rooms are usually involved in the design of a pharmaceutical production facility. The term ‘cleanroom’ is specified in detail in the ISO standards 14644 (parts 1, 2, 4, 5 and 7) and 14698 (parts 1 and 2) [2–5]. Particularly ISO 14644 part 4 deals with the design, the construction and the initial start-up of a clean room and includes a useful checklist [4].
27.4 Functional Specification
27.4.1 Contents
Next step in the design process is drawing the functional requirements specification (FRS), also called the physical requirements specification. The FRS documents elaborated demands for connections, heat burden, floor load, acoustic demands, specifications of the walls, HVAC etc.
The heat burden is the amount of heat that is generated in a room per unit of time by humans and apparatus. Provisions for air supply have direct impact on product quality. In the FRS for clean rooms the limits are specified for the allowable number of particles at rest and in operation, i.e. the clean room class (see Sect. 27.4.2 below), along with desired turbulence limits, limits for air pressure and microbiological limits. It provides the final specifications for premises, fixtures and fittings and a map with the positions of furniture and apparatus. The FRS contains also technical or procedural measures to prevent cross contamination or crossing lines for personal or goods [5]. Obviously all specifications in the FRS are substantiated at the base of their application. As an example, in premises intended for complex aseptic handling or reconstitution of hazardous sterile medicines, any choice for the classification of the background conditions should be justified.
27.4.2 Classification of Premises
The idea behind GMP is to preclude any factor with an unpredictable or undetermined impact on the production process as this impairs the process validity. Particles, micro-organisms and (gaseous) chemical contaminants arising from the air from outside might constitute such a factor, especially relevant for the process of producing sterile and aseptic preparations. Therefore, GMP requires that premises for this kind of preparations must be classified. Since the absence of invisible micro-organisms and dust is hard to be proved at any moment and place, measures are prescribed that should guarantee this absence with substantial safety margins:
The inlet air should be filtered, through HEPA (Highly Efficient Particulate Air) filters.
A specified air replacement factor per hour (ventilation factor; see Sect. 27.5.1 HVAC) should be achieved.
The premises must be kept clean in relation to lower classified neighbouring premises by the application of pressure differences.
Annex 1 of the GMP applies. This document specifies four grades for low particulate premises: A, B, C and D. See Table 27.2.
GMP Annex 1 states that a certain air quality class can only exist when the access to the room is achieved through an air lock in which the same class prevails. The Annex 1 classification refers to the ISO classification (see Table 27.3) that define intermediate air quality specifications by decimal class designations.
Table 27.3
ISO 14644–1 classification of air cleanliness
Class | Maximum permitted number of particles per m3 equal to or greater than the tabulated size (in operation) | |||||
|---|---|---|---|---|---|---|
≥0.1 μm | ≥0.2 μm | ≥0.3 μm | ≥0.5 μm | ≥1 μm | ≥5 μm | |
1 | 10 | 2 | ||||
2 | 100 | 24 | 10 | 4 | ||
3 | 1,000 | 237 | 102 | 35 | 8 | |
4a | 10,000 | 2,370 | 1,020 | 352 | 83 | |
5(GMP B at rest) | 100,000 | 23,700 | 10,200 | 3,520 | 832 | 29 |
6 | 1,000,000 | 237,000 | 102,000 | 35,200 | 8,320 | 293 |
7 (GMP grade B in operation, Grade C at rest) | 352,000 | 83,200 | 2,930 | |||
8 (GMP grade C in operation, Grade D at rest) | 3,520,000 | 832,000 | 29,300 | |||
9 | 35,200,000 | 8,320,000 | 293,000 | |||
According to ISO 14644–1 the air classification scheme is distinguished by a mathematically coherent approach and based upon a formula:
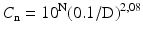
where
(29.1)
Cn = maximum number concentration of particles per m3 with diameter ≥ the considered particle diameter, rounded to a maximum of 3 digits;
N = ISO classification number;
D = considered particle diameter;
0.1 = the reference diameter, a constant with the dimension mm.
As the ISO classification is defined by a continuous formula with the particle size as a variable and the GMP grades are defined in discrete counts for specific particle sizes, the comparison between GMP grades and ISO classifications is formally not possible. Additionally ISO classifications can be formulated in decimals where the GMP grades cannot. However as a practical approach the comparison is possible as e.g. grade B at rest will match ISO 5 for most practical purposes.
How Is the Classification of the Premises Actually to be Derived from the URS?
A basic principle is that raw materials, intermediate products and final products that are not yet in their final, primary package should never be exposed to any unconditioned air. The primary package is the airtight enclosing package of the final product that is in direct contact with the product. Should therefore also non-sterile extemporaneous preparations (Category I; community pharmacy or small hospital pharmacy) be prepared in a classified premise? The principal objective is that during preparation of non-sterile products no contamination from whatever source can occur. To that end adequate procedures should be organised such as a warranted adequate maintenance (cleaning, prevention of impairment) and also warranted procedures should apply to prevent disturbances such as any opening of doors or windows during preparation activities. Under such conditions, a specific classification of premises has limited meaning and usually can be left out. After all Class D only specifies a maximum number of particles at rest which is usually easy achievable except when a current badly functioning ventilation system is in action or during specific (weather) conditions, incurring a high concentration of fine particles or pollen.
Also if products are sterilised in their primary package a low microbial starting contamination is essential. Therefore products to be sterilised must be filled under class C conditions but the preparation of the bulk solution may take place under class D conditions.
Aseptic handling and preparation must be executed under class A conditions, usually a LAF (Laminar Air Flow) cabinet, a LDF (Laminar Down Flow) cabinet, a safety cabinet or an isolator see Sect. 28.3. The background room for stock preparations in a LAF, or LDF cabinet is at least class B. The background room for aseptic reconstitution or handling in a LAF, or safety cabinet may have a lower classification than B, provided that it is based on risk assessment. Conformation to the latest views of inspectorates and professional associations e.g. PIC/S [1] is advised.
For premises designed for non-sterile preparations overpressure is preferable to prevent any penetration of uncontrolled air. For premises designed for sterile or aseptic preparations this is required by GMP.
Generally working with hazardous substances – as almost all active substances are – requires containment. In the specific case of radiopharmaceuticals, Nuclear Energy legislation requires the application of negative pressure (see Sect. 15.6.3). In the situation of aseptic handling, which requires positive pressure, with radiopharmaceuticals, this results essentially in contradictory pressure demands. A solution may be achieved by putting the whole complex of preparation rooms with air locks at negative pressure relative to its environment. The system of walls, floors and ceilings should be completely airtight in that case. Pipe entries, electrical sockets as well as porous stone in the wall parts above the ceiling are frequent sources of (substantial) leakage of contaminated air. Thus all chinks and gaps have to be filled and porous stone has to be coated.
By now putting the air locks at some additional negative pressure a relative overpressure in the preparation premises is created relative to the admission air locks. The deeper negative pressure in the air lock will however lead to a vigorous influx of contaminated air as soon as the outer door is opened, contaminating the lock and compromising its function. To prevent this an additional ‘front air lock’ should be considered. The front air lock should have a slight negative pressure relative to the environment and will be supplied with clean HEPA filtered air. Alternatively, a clean air lock or corridor giving access to other (positive pressure) premises can serve as a front air lock to a negative pressure premise. See Fig. 27.1.
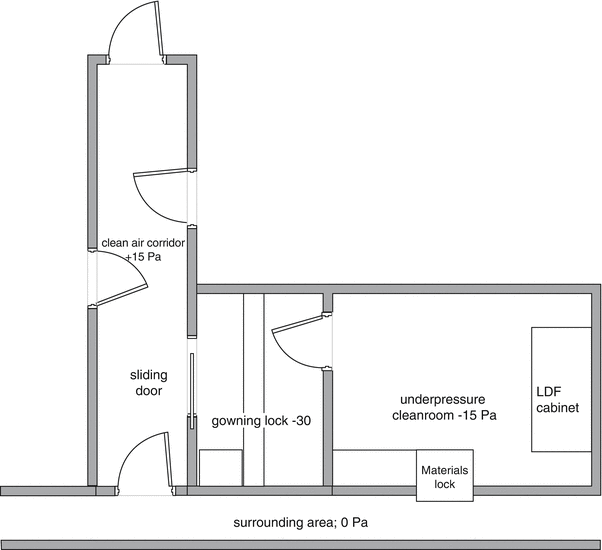
Fig. 27.1
Model plan for pressure hierarchy around negative pressure premises. Adapted from Recepteerkunde 2009, ©KNMP
In many respects the hazards of many antineoplastics and radiopharmaceuticals for staff are similar. Also the required skills for aseptic handling are similar. This may lead to the consideration of designing a standard layout for all premises for handling with extremely hazardous substances. However, for antineoplastics no formal requirement for pressure applies (see Sect. 26.8). Therefore, the viewpoint that overpressure warrants a better microbiological air quality usually will prevail. In addition the validity of negative pressure premises may be impaired as a result of almost unavoidable air leakages.
The objective can also be achieved with the use of negative pressure cabinets sited in positive pressure rooms.
Weighing of solid substances will release dust. A dust extract cabinet, equipped with a High Efficiency Particulate Air (HEPA) or Ultra Low Particulate Air (ULPA) filter (see Table 27.4) in the rear wall or in the exhaust, or both, will limit the exposure of operators to active substances. Additional options are the wearing of respirators (see Sect. 26.4.1) and a closed weighing vessel.
Table 27.4
Survey of HEPA and ULPA filters
Filter class according to EN 1822:2009 | Efficiency % | Penetration % |
|---|---|---|
E10 | 85 | 15 |
E11 | 95 | 5 |
E12 | 99.5 | 0.5 |
H13 | 99.95 | 0.05 |
H14 | 99.995 | 0.005 |
U15 | 99.9995 | 0.0005 |
U16 | 99.99995 | 0.00005 |
U17 | 99.999995 | 0.000005 |
The air turbulence from a dust extract cabinet may disturb the functioning of electronic balances. A dedicated construction in each of the preparation premises to manage this is expensive and therefore cannot always be achieved. If for this reason a separate weighing chamber is designed all measured portions should be transported in well-closed and identified vessels.
27.4.3 Interlock Systems for Air Locks
For the maintenance of an air pressure regime and an air quality class of a preparation premise an air lock is required. It controls access from any lower qualified premise. Personnel locks should be discriminated from materials locks. In personnel locks the required gowning regime has to be determined in advance. In most cases this results in the requirement of sex-separated gowning locks.
For materials locks the prevailing transport direction and the usual volume of materials is important. If possible the transport direction should be assured for instance by means of a catch that transmits a standardised vehicle (e.g. a crate) in only one direction.
All air locks must be equipped with a so-called interlock system. This system ensures that both doors of the air lock will never be opened at the same time.
Different interlock principles exist:
Both doors are permanently closed by magnets, necessitating a separate hand operated switch to open one of them.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


