Meningitis-associated Escherichia coli
Kwang Sik Kim, Johns Hopkins University School of Medicine, Baltimore, MD, USA
Introduction
Gram-negative bacillary meningitis continues to be an important cause of mortality and morbidity throughout the world. Case fatality rates have ranged between 15% and 40%, and approximately 50% of survivors sustain neurological sequelae (Gladstone et al., 1990; Unhanand et al., 1993; Dawson et al., 1999; Klinger et al., 2000; Stevens et al., 2003). A major contributing factor to such mortality and morbidity is our incomplete understanding of the pathogenesis of this disease (Kim, 2001, 2002, 2003, 2008, 2010, 2012). Both clinical and experimental data indicate limited efficacy with antimicrobial therapy alone for the treatment of Gram-negative bacillary meningitis (McCracken et al., 1984; Kim, 1985). E. coli is the most common Gram-negative organism that causes meningitis, particularly during the neonatal period.
An emergence of antibiotic resistance is an additional contributing factor to mortality and morbidity associated with E. coli meningitis. Recent reports of neonatal meningitis caused by E. coli strains producing CTX-M-type or TEM-type extended-spectrum β-lactamases are a particular concern (Blanco et al., 2011; Moissenet et al., 2011). Drug-resistant clonal group ST131 is prevalent among E. coli strains causing extraintestinal infection and some carry CTX-M-type extended-spectrum β-lactamases (Blanco et al., 2011). These findings indicate that a novel strategy is needed to identify new targets for prevention and therapy of E. coli meningitis.
Several lines of evidence from human cases of E. coli meningitis and animal models of experimental hematogenous E. coli meningitis indicate that E. coli invasion into the brain follows a high level of bacteremia and cerebral capillaries are the portal of entry into the brain (Berman and Banker, 1966; Kim et al., 1992), but how meningitis-causing E. coli strains invade the blood–brain barrier and penetrate into the brain remains incompletely understood.
Given the plethora of E. coli serotypes, it is striking that E. coli strains possessing the K1 capsular polysaccharide are predominant (approximately 80%) among isolates from neonatal E. coli meningitis (Robbins et al., 1974; Gross et al., 1982; Korhonen et al., 1985) and most of these K1 isolates are associated with a limited number of O serotypes (e.g. O18, O7, O16, O1, O45), belonging to phylogenetic group B2 and to a lesser extent, to group D (Sarff et al., 1975; Bingen et al., 1998; Johnson et al., 2001; Bonacorsi et al., 2003). The basis of this association of phylogenetic groups B2 and D with E. coli meningitis has not been elucidated.
The development of both in vitro and in vivo models of the blood–brain barrier has facilitated our investigations on the mechanisms of the microbial traversal of the blood–brain barrier, a key step required for the development of E. coli meningitis (Kim, 2001, 2002, 2003, 2008, 2010, 2012). The blood–brain barrier protects the brain from microbes circulating in the blood, but meningitis-causing pathogens have been shown to traverse the blood–brain barrier transcellularly, paracellularly and/or by means of infected phagocytic cells (Trojan-horse mechanism) (Kim, 2008). Recent studies have demonstrated that meningitis-causing E. coli K1 strains exhibit the ability to traverse the blood–brain barrier by transcellular penetration and cause central nervous system (CNS) inflammation, resulting in meningitis (Kim, 2001, 2002, 2003, 2008, 2010, 2012).
E. coli traversal of the blood–brain barrier
The blood–brain barrier is a structural and functional barrier that is formed by brain microvascular endothelial cells (BMEC), astrocytes, and pericytes (Rubin and Staddon, 1999). It regulates the passage of molecules into and out of the brain to maintain the neural microenvironment, and astrocytes and pericytes help maintain the barrier property of BMEC. The contributions of astrocytes and pericytes to E. coli traversal of the blood–brain barrier are, however, shown to be minimal.
The in vitro blood–brain barrier model has been developed with human brain microvascular endothelial cells (HBMEC). Upon cultivation on collagen-coated Transwell inserts these HBMEC exhibit morphologic and functional properties of tight junction formation and form a polarized monolayer. These properties are shown by the demonstrations of tight junction proteins (such as claudin 5 and ZO-1) and adherens junction proteins (such as VE-cadherin and β-catenin) and their spatial separation, as well as development of high transendothelial electrical resistance (Stins et al., 1997, 2001; Kim et al., 2004; Ruffer et al., 2004). Our previous studies with scanning and transmission electron microscopy documented the internalization of meningitis-causing E. coli K1 strains into HBMEC, as shown by the demonstration that internalized bacteria are found within membrane-bound vacuoles of HBMEC (Figure 10.1). E. coli K1 transmigrates the HBMEC monolayer through an enclosed vacuole without intracellular multiplication and without any change in the integrity of the HBMEC monolayer (Nemani et al., 1999; Stins et al., 2001; Kim, 2003). No free bacteria are found in the cytoplasm of HBMEC or between adjacent HBMEC.
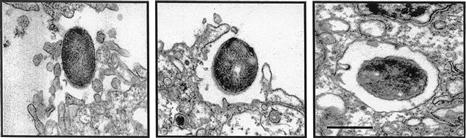
FIGURE 10.1 Transmission electron micrograph of human brain microvascular endothelial cell monolayer infected with meningitis-causing E. coli K1 strain RS218 (018:K1, B2 group). Cellular protrusions surround internalizing E. coli (left) and internalized E. coli are found within membrane-bound vacuoles (right). Scale bar = 1 µm. (Adapted from Kim, 2003, figure 3.)
The in vivo blood–brain barrier model has been developed by inducing hematogenous meningitis in infant rats and mice. In this animal model, E. coli is administered orally, subcutaneously, intracardially, or intravenously, resulting in bacteremia and subsequent entry into the CNS (Kim et al., 1992; Huang et al., 1995, 1999; Wang et al., 1999; Hoffman et al., 2000; Khan et al., 2002; Wang and Kim, 2002; Wang et al., 2004; Zhu et al., 2010a,b). Studies in experimental hematogenous meningitis models indicate that the primary site of entry into the CNS for circulating E. coli is the cerebral vasculature, not the choroid plexus (Kim et al., 1992). E. coli entry into the CNS was documented without any change in the blood–brain barrier permeability as well as without concomitant presence of host inflammatory cells (e.g. PMNs, macrophages) (Kim et al., 1992, 1997), excluding the possibility that E. coli penetrates into the brain using the paracellular mechanism or the Trojan-horse mechanism via transmigration of E. coli-infected phagocytic cells. Taken together, these findings indicate a transcellular penetration of meningitis-causing E. coli K1 across the blood–brain barrier.
It is important to point out that E. coli is an enteric organism and there are no data supporting the concept that E. coli meningitis occurs following inhalation or intranasal spread (Mittal et al., 2010) and also there are no data indicating that an exposure to passive smoking (e.g. nicotine) increases the development of E. coli meningitis (Chi et al., 2011).
Studies with the above-mentioned in vitro and in vivo models of the blood–brain barrier have demonstrated that successful traversal of the blood–brain barrier by circulating E. coli requires (a) a high degree of bacteremia; (b) E. coli binding to and invasion of HBMEC; and (c) traversal of the blood–brain barrier as live bacteria (Kim et al., 1992; Hoffman et al., 1999; Kim 2001, 2002, 2003, 2008, 2010, 2012) (Table 10.1).
A threshold level of bacteremia required for E. coli penetration into the brain
Several studies in humans and experimental animals point to a relationship between the magnitude of bacteremia and the development of meningitis due to E. coli. For example, a significantly higher incidence of E. coli meningitis was noted in neonates who had bacterial counts in blood higher than 103 colony-forming units (CFU)/ml (6 out of 11 or 55%), compared to those with blood bacterial counts lower than 103 CFU/ml (1 out of 19 or 5%) (Dietzman et al., 1974). A high degree of bacteremia was also shown to be a primary determinant for penetration into the brain by circulating E. coli K1 in neonatal and adult animals with experimental hematogenous E. coli meningitis (Kim et al., 1992; Huang et al., 1995, 1999; Wang et al., 1999, 2004; Hoffman et al., 2000; Khan et al., 2002; Wang and Kim, 2002), but an approximately 106-fold greater inoculum of E. coli K1 is required to induce a similar high-level bacteremia in adult animals compared to neonatal animals (Kim et al., 1992). These findings suggest that the age dependency of E. coli meningitis is most likely due to the relative resistance of adults to high-level bacteremia, which precedes the development of meningitis, and less likely due to greater invasion of meningitis-causing E. coli in HBMEC derived from neonates compared to those from adults. This concept is shown by our demonstration that the abilities of meningitis-causing E. coli K1 to bind and invade BMEC are similar between BMEC derived from young and old rats as well as HBMEC derived from different ages (Stins et al., 1999). Thus, one of the reasons for the close association of meningitis-causing E. coli strains with neonatal meningitis is their ability to escape from host defenses and then to achieve a threshold level of bacteremia necessary for invasion of the meninges. Taken together, these findings indicate that the prevention of bacterial multiplication in the blood that is required for penetration into the brain would be one potential approach for prevention of E. coli meningitis.
Previous studies have identified that the expression of K1 capsular polysaccharide and O-lipopolysaccharide (LPS) are shown to be critical for induction of a high degree of bacteremia (Cross et al., 1986; Kim et al., 1986, 1988), but the feasibility of using the K1 capsule and O-LPS for the prevention of E. coli bacteremia has been shown to be limited (Soderstrom et al., 1984; Finne et al., 1987; Cross et al., 1994).
Recent functional E. coli genomic studies identified several E. coli factors that are shown to contribute to bacteremia (Kim, 2008; Xie et al., 2008; Moriel et al., 2010). For example, NlpI is a lipoprotein located in the outer membrane and has been shown to contribute to a high-level E. coli K1 bacteremeia. NlpI’s evasion of serum-mediated killing is through regulation of complement regulator C4bp deposition on the bacterial surface (Tseng et al., 2012). Studies are in progress to determine the protective and broadly conserved antigens or to develop a multi-epitope subunit vaccine for the prevention of E. coli bacteremia and subsequent meningitis (Moriel et al., 2010; Wieser et al., 2012).
E. coli binding to and invasion of HBMEC
Subsequent studies have shown that a high degree of bacteremia is necessary, but not sufficient for E. coli penetration of the blood–brain barrier in vivo, and that E. coli binding to and invasion of HBMEC is a prerequisite for penetration into the brain (Huang et al., 1995, 1999; Wang et al., 1999; Hoffman et al., 2000; Khan et al., 2002; Wang and Kim, 2002), the essential step for the development of E. coli meningitis. This was shown by the demonstration in infant rats with experimental hematogenous meningitis that several isogenic mutants of meningitis-causing E. coli K1 strain RS218 deleted of factors contributing to HBMEC binding (e.g. OmpA) and invasion (e.g. Ibe proteins, CNF1) were significantly less able to induce meningitis than the parent strain despite similar levels of bacteremia (Table 10.2). These findings indicate that those E. coli factors contributing to HBMEC binding and invasion are necessary for crossing the blood–brain barrier in vivo.
TABLE 10.2
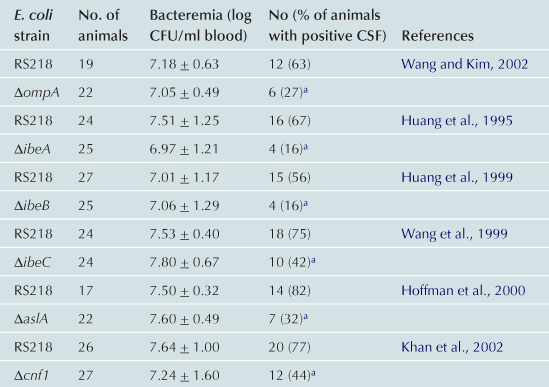
aSignificantly less than RS218.
Modified with permission from Infection and Immunity (Kim, 2001).
E. coli factors contributing to HBMEC binding
Infections caused by pathogenic E. coli are often initiated by binding of the bacteria to the host cell surface, and this concept is likely to be important for circulating E. coli to withstand the blood flow in vivo and cross the blood–brain barrier. Several E. coli factors are identified to be involved in binding to HBMEC that subsequently affect invasion into HBMEC. These bacterial factors include type 1 fimbriae (FimH), flagella (FliC), outer membrane protein A (OmpA) and a lipoprotein (NlpI) (Khan et al., 2003, 2007; Shin et al., 2005; Teng et al., 2005, 2010; Parthasarathy et al., 2007). The roles of these E. coli factors in HBMEC binding were verified by deletion and complementation experiments. For example, isogenic deletion mutants were significantly less able to bind HBMEC and their binding ability was restored to the level of the parent strain by complementation with respective wild-type genes.
Fimbriae
Our experiment with an E. coli DNA microarray comparing the gene expression patterns of HBMEC-associated versus non-associated E.coli K1 revealed that type 1 fimbriae play an important role in E. coli K1 binding to HBMEC (Teng et al., 2005). The HBMEC-associated E. coli K1 showed significantly higher expression levels of the fim cluster genes compared to non-associated bacteria. Expression of type 1 fimbriae in wild-type E. coli is regulated by phase variation in which each bacterium can alternate between fimbriated and non-fimbriated states, so-called phase-ON and phase-OFF, respectively (see Chapter 12). We have shown that E. coli K1 associated with HBMEC are predominantly type 1 fimbriae phase-ON bacteria. We constructed type 1 fimbria locked-ON and locked-OFF mutants of E. coli K1 strain RS218, whose fim promoters are fixed in the ON and OFF orientations, respectively, and showed that E. coli K1 binding to HBMEC is significantly greater with the locked-ON mutant than the wild-type strain, while it is significantly less with the locked-OFF mutant (Teng et al., 2005). Decreased binding as the result of the fimH deletion or the locked-OFF mutant resulted in decreased invasion into HBMEC (Teng et al., 2005; Khan et al., 2007).
From our E. coli DNA microarray experiments, we identified a novel site-specific recombinase, HbiF, which inverted the molecular switch fimS independent of the two known recombinases, FimB and FimE, that invert fimS and control the expression of the downstream fim operon (Xie et al., 2006a,b). Discovery of HbiF-mediated fimS switching provides a new opportunity for regulating type 1 fimbriae expression, which will help in developing a novel strategy for the prevention and therapy of E. coli bacteremia and meningitis.
We also identified that FimH interacts with a glycosylphosphatidylinositol-anchored receptor, CD48 on the surface of HBMEC, and FimH-CD48 interaction contributes to E. coli binding to HBMEC and increases in intracellular Ca2+ ([Ca2+]i) in HBMEC (Khan et al., 2007; Kim et al., 2008). This concept is shown by the demonstration that CD48 antibody blocks FimH-mediated binding to HBMEC and FimH-induced [Ca2+]i changes in HBMEC (Table 10.3).
S fimbriae, which bind to terminal NeuAc α2,3-galactose sequences present on glycoproteins, have been implicated in E. coli binding to HBMEC. For example, purified S fimbriae or a recombinant E. coli strain HB101 expressing S fimbriae was shown to bind to the luminal surfaces of the brain vascular endothelium in neonatal rat brain tissues (Parkkinen et al., 1988). We have previously shown using S fimbriated transformants of E. coli strain HB101 that S fimbriae allowed this laboratory strain of E. coli to bind to HBMEC (Prasadarao et al., 1993; Stins et al., 1994), suggesting that S fimbriae contribute to E. coli binding to HBMEC. However, in-frame deletion of the S fimbriae operon in meningitis-causing E. coli K1 did not significantly affect E. coli binding to and invasion of HBMEC, and also did not affect E. coli K1 penetration into the brain in the experimental hematogenous meningitis animal model (Wang et al., 2004). These findings suggest that S fimbriae are not critical to HBMEC binding in vitro and traversal of the blood–brain barrier in vivo by meningitis-causing E. coli K1.
Flagella
The external whip-like structures involved in bacterial motility have been shown to contribute to the virulence of many enteric bacteria, including E. coli (Parthasarathy et al., 2007). Our experiment with E. coli DNA microarray comparing the gene expression patterns of HBMEC-associated versus non-associated E.coli K1 also demonstrated that flagella play an important role in E. coli K1 binding to HBMEC (Parthasarathy et al., 2007; Xie et al., 2008). The flagellum is synthesized from three classes of genes. The class I region consists of the master operon flhDC, the class II region comprises the genes involved in the synthesis of the flagella basal body and hook, and the class III region consists of genes involved in flagella synthesis and motility. Using defined deletion mutants constructed for each class of genes, we showed that the mutants deleted of the flagella structure exhibited significant defects in HBMEC binding and invasion, while deletion of a gene affecting only motility had no defects in HBMEC binding and invasion (Parthasarathy et al., 2007). These findings indicate that bacterial flagellin plays an important role in meningitis-causing E. coli K1 binding to and invasion of HBMEC. This concept is supported by the demonstration that recombinant flagellin (FliC) binds directly to the surface of HBMEC and exogenous recombinant flagellin decreases E. coli K1 binding to HBMEC (Parthasarathy et al., 2007).
OmpA
OmpA is one of the major outer membrane proteins in E. coli and its N-terminal domain crosses the outer membrane eight times in antiparallel β-strands with four hydrophilic surface-exposed loops and short periplasmic turns. We have shown that the N-terminal portion of OmpA and its surface-exposed loops contribute to binding to HBMEC (Nemani et al., 1996a,b; Shin et al., 2005; Maruvada and Kim, 2011). We also showed that OmpA interacts with HBMEC through N-acetylglucosamine (GlcNAc) residues of gp 96 (Nemani et al., 1996b; Khan et al., 2003). The chitooligomers (GlcNAc β1, 4-GlcNAc oligomers) and chitohexose block meningitis-causing E. coli K1 invasion of HBMEC and traversal of the blood–brain barrier in the infant rat model of experimental hematogenous meningitis (Nemani et al., 1996b; Maruvada and Kim, 2011).
Our recent study with an E. coli DNA microarray comparing the ompA deletion mutant with its parent E. coli K1 strain RS218, however, revealed that the ompA deletion mutant exhibited significantly lower expression of the fim cluster genes, and lower expression of type 1 fimbriae on the bacterial surface (Teng et al., 2006). These findings suggest that decreased binding of the ompA deletion mutant may be related to its lower expression of type 1 fimbriae. The ompA deletion mutant was significantly less efficient in its penetration into the brain in vivo compared to the parent E. coli K1 strain (Wang and Kim, 2002). Additional studies are needed to determine whether these in vitro and in vivo defects of the ompA deletion mutant are in part related to its decreased expression of type 1 fimbriae and to also understand how the deletion of ompA affects type 1 fimbria expression.
NlpI
NlpI is shown to be an important factor of Crohn’s disease-associated E. coli strain LF82 (083:H1) to interact with intestinal epithelial cells (Barnich et al., 2004). Deletion of nlpI in E. coli strain LF82 decreased expression of type 1 fimbriae and flagella (Barnich et al., 2004). We showed that NlpI is an outer membrane-anchored protein and contributes to meningitis-causing E. coli K1 binding to and invasion of HBMEC (Teng et al., 2010). Unlike strain LF82, deletion of nlpI in meningitis-causing E. coli, however, did not affect the expression of type 1 fimbriae, flagella and OmpA, indicating that the contribution of NlpI to HBMEC binding and invasion is independent of those bacterial factors in meningitis-causing E. coli. This concept is shown by the demonstration that mutants deleted of type 1 fimbriae, OmpA and NlpI exhibited significantly decreased HBMEC binding and invasion compared to mutants deleted of individual factors or a combination of the two factors (Teng et al., 2010). These findings suggest that type 1 fimbriae, OmpA and NlpI are likely to contribute to HBMEC binding and invasion independent of each other. It remains, however, incompletely understood how and why several bacterial factors of meningitis-causing E. coli are involved in HBMEC binding.
E. coli structures contributing to invasion of HBMEC
Previous studies using TnphoA mutagenesis, signature-tagged mutagenesis and differential fluorescence induction with screening of a gfp fusion library identified several E. coli determinants contributing to invasion of HBMEC, which include Ibe (named after invasion of brain endothelial cell) proteins and cytotoxic necrotizing factor 1 (CNF1) (Huang et al., 1995, 1999; Wang et al., 1999; Badger et al., 2000a,b; Hoffman et al., 2000; Khan et al., 2002). Isogenic deletion mutants were significantly less invasive in HBMEC and less able to penetrate into the brain in vivo (Table 10.2) and their invasive defects were restored to the levels of the parent strain by complementation with respective wild-type genes.
Ibe proteins
Ibe A, B, and C proteins were identified via TnphoA mutagenesis of meningitis-causing E. coli K1 strain RS218 and screening of the mutants for loss of invasion by use of both the in vitro and in vivo blood–brain barrier models (Huang et al., 1995, 1999; Wang et al., 1999).
Exogenous recombinant Ibe proteins were shown to inhibit meningitis-causing E. coli K1 invasion of HBMEC (Huang et al., 1995), suggesting that Ibe proteins contribute to HBMEC invasion by ligand–receptor interactions. This concept was supported by the demonstration of a HBMEC surface protein interactive with IbeA, and a polyclonal antibody raised against this receptor protein inhibited E. coli K1 invasion of HBMEC (Kim, 2001). The gene, ibeA, was shown to be prevalent in meningitis-causing E. coli K1 of phylogenetic B2 group (Johnson et al., 2002; Bonacorsi et al., 2003), while ibeB and ibeC were found to have homologs in non-meningitis isolates of E. coli. It remains unclear whether the ibeB and ibeC homologs from non-meningitis isolates will exhibit the HBMEC invasion phenotype similar to that of meningitis-causing E. coli K1 strains. The mechanisms involved with IbeA, IbeB, and IbeC in their contributions to E. coli K1 invasion of HBMEC, however, appear to be similar and their contributions to HBMEC invasion are likely to be redundant. This is shown by the demonstration that HBMEC invasion frequencies did not differ significantly between mutants deleted of ibeA, ibeB, and ibeC versus mutants deleted of single or double genes (Kim, 2002). Additional studies are needed to elucidate the mechanisms involved with IbeA, IbeB, and IbeC for their contribution to E. coli meningitis.
CNF1
CNF1 is a bacterial virulence factor associated with extraintestinal pathogenic E. coli strains causing urinary tract infection and meningitis (Bouquet, 2001). CNF1 is an AB-type toxin, composed of the N-terminal cell binding domain and the C-terminal catalytic domain possessing a deaminase activity through the site-specific deamination of a Gln residue to Glu (Flatau et al., 1997; Schmidt et al., 1997). CNF1 has been shown to activate RhoGTPases such as RhoA and induce uptake of latex beads, bacteria, and apoptotic bodies into non-professional phagocytes such as epithelial and endothelial cells by macropinocytosis (Fabbri et al., 2002).
CNF1 contributes to E. coli K1 invasion of HBMEC in vitro and traversal of the blood–brain barrier in vivo, and these in vitro and in vivo effects of CNF1 are dependent upon RhoA activation (Khan et al., 2002). These conclusions were shown by (a) significantly decreased invasion and RhoA activation with the CNF1 deletion mutant compared to the parent strain in HBMEC and (b) restoration of the CNF1 mutant’s invasion frequency to the level of the parent strain in HBMEC expressing constitutively active RhoA.
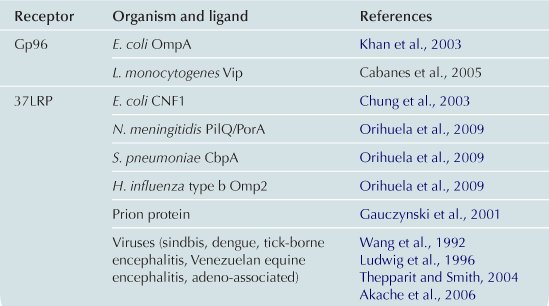
CNF1 has been suggested to be internalized via receptor-mediated endocytosis upon binding to a cell surface receptor (Bouquet, 2001). We have identified the HBMEC receptor for CNF1 by yeast two-hybrid screening of the HBMEC cDNA library using the N-terminal cell-binding domain of CNF1 as bait (Chung et al., 2003). This receptor, 37-kDa laminin receptor precursor (LRP) interacted with the N-terminal CNF1 and full-length CNF1 but not with the C-terminal CNF1. CNF1-mediated RhoA activation and bacterial uptake were inhibited by LRP antisense oligodeoxynucleotides,whereas they were increased in LRP-overexpressing cells, demonstrating correlation between effects of CNF1 and levels of LRP expression in HBMEC (Chung et al., 2003). These findings indicate that CNF1 interaction with its receptor, LRP, is the initial step required for CNF1-mediated RhoA activation and bacterial uptake in eukaryotic cells. The 37-kDa LRP is a ribosome-associated cytoplasmic protein and shown to be a precursor of 67-kDa laminin receptor (67 LR). It is unclear how 67 LR is matured and synthesized from the LRP, but mature 67 LR is shown to be present on the cell surface and functions as a membrane receptor for the adhesive basement membrane protein laminin (Massia et al., 1993). We have shown that incubation of HBMEC with CNF1-expressing E. coli K1 up-regulates 67 LR expression and recruits 67 LR to the site of invading E. coli K1 in a CNF1-dependent manner (Kim et al., 2003), supporting the participation of 67 LR in CNF1-expressing E. coli K1 invasion of HBMEC.
Of interest, LRP is also shown to be a cellular target for other CNS-infecting microorganisms, including S. pneumoniae, N. meningitidis, H. influenzae type b, dengue virus, adeno-associated virus, Venezuelan equine encephalitis virus, and prion protein (Table 10.3). The mechanisms by which the same receptor is involved in CNS penetration by different organisms remain to be established.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


