FIGURE 15.1 Chemical structures of lipids. Fatty acids are abbreviated as (R) for triglycerides and phospholipids.
The polyunsaturated fatty acids are classified into omega-3, omega-6, and omega-9 families, depending on the position of the first double bond from the terminal (omega) methyl group of the fatty acid chain. Polyunsaturated fatty acids are precursors for the synthesis of eicosanoids that include prostaglandins, thromboxanes, prostacyclins, and leukotrienes. These fatty acids play a vital role in the structure and function of most biological membranes, and some fatty acids like omega-3 polyunsaturated fatty acids found in fish oils are beneficial in lowering the risk of cardiovascular disease.
Triglycerides
As can be inferred from the name, triglycerides contain three fatty acid molecules attached to one molecule of glycerol by ester bonds in one of three stereochemically distinct bonding positions, referred to as sn-1, sn-2, and sn-3 (Fig. 15.1). Each fatty acid in the triglyceride molecule can potentially be different in structure, thus producing many possible types of triglycerides. Triglycerides containing saturated fatty acids, which do not have bends in their structure (Fig. 15.1), pack together more closely and tend to be solid at room temperature. In contrast, triglycerides, containing cis unsaturated fatty acids (Fig. 15.1), typically form oils at room temperature. Most triglycerides from plant sources, such as corn, sunflower seeds, and safflower seeds, are rich in polyunsaturated fatty acids and are oils, whereas triglycerides from animal sources contain mostly saturated fatty acids and are usually solid at room temperature. As can be seen by inspecting the structure of triglycerides (Fig. 15.1), there are no charged or polar hydrophilic groups, making it very hydrophobic and virtually water insoluble. Because it is uncharged, triglyceride is classified as a neutral lipid.
Phospholipids
Phospholipids are similar in structure to triglycerides except that they only have two esterified fatty acids (Fig. 15.1). The third position on the glycerol backbone instead contains a phospholipid head group. There are several types of phospholipid head groups, such as choline, inositol, inositol phosphates, glycerol, serine, and ethanolamine, which are all hydrophilic in nature. The various types of phospholipids are named based on the type of phospholipid head group present. Phosphatidylcholine (often referred as lecithin) (Fig. 15.1) has a choline head group and is the most common phospholipid found on lipoproteins and in cell membranes. The two asymmetrically positioned fatty acids in phospholipids are typically 14 to 24 carbon atoms long, with one fatty acid commonly saturated and the other unsaturated most commonly in the sn-2 position.
Because phospholipids contain both hydrophobic fatty acid C–H chains and a hydrophilic head group, they are by definition amphipathic lipid molecules and, as such, are found on the surface of lipid layers or on the surface of lipoprotein particles. The polar hydrophilic head group faces outward toward the aqueous environment, whereas the fatty acid chains face inward away from the water in a perpendicular orientation with respect to the lipid surface. Phospholipids are synthesized in the cytosolic compartment of all organs of the body, especially in the liver, with phosphatidylcholine and phosphatidylethanolamine being the most abundant phospholipids in the body.
Cholesterol
Cholesterol is an unsaturated steroid alcohol containing four rings (A, B, C, and D), and it has a single C–H side chain tail similar to a fatty acid in its physical properties (Fig. 15.1). The only hydrophilic part of cholesterol is the hydroxyl group in the A-ring. Cholesterol is, therefore, also an amphipathic lipid and is found on the surface of lipid layers along with phospholipids. Cholesterol is oriented in lipid layers so that the four rings and the side chain tail are buried in the membrane in a parallel orientation to the fatty acid acyl chains on adjacent phospholipid molecules. The polar hydroxyl group on the cholesterol A-ring faces outward, away from the lipid layer, allowing it to interact with water by noncovalent hydrogen bonding or with the polar head groups of phospholipids.
Cholesterol can also exist in an esterified form called cholesteryl ester, with the hydroxyl group conjugated by an ester bond to a fatty acid, in the same way as in triglycerides. In contrast to free cholesterol, there are no polar groups on cholesteryl esters, making them very hydrophobic. Because it is not charged, cholesteryl esters are also classified as a neutral lipid and are not found on the surface of lipid layers but instead are located in the center of intracellular lipid droplets or in the hydrophobic core of lipoproteins, along with triglycerides.
Cholesterol is almost exclusively synthesized by animals, but plants do contain other sterols (called phytosterols) similar in structure to cholesterol. Dietary phytosterols are known to lower plasma total cholesterol and LDL cholesterol (LDL-C) and raise HDL cholesterol (HDL-C) levels most likely by competing with the intestinal absorption of cholesterol.
Cholesterol is synthesized in most tissues of the body from acetyl-CoA in the microsomal and cytosolic compartments of the cell. More than 25 enzymes are involved in the formation of cholesterol from acetyl-CoA. The principal steps include the conversion of acetyl-CoA derived from either the β-oxidation of fatty acids or the oxidative decarboxylation of pyruvate to β-hydroxy β-methyl glutaryl CoA (HMG-CoA).3 HMG-CoA is then converted to mevalonic acid by the enzyme HMG-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis.3 Mevalonic acid is phosphorylated, isomerized, and converted to geranyl- and farnesyl pyrophosphate, which, in turn, forms squalene.3 Cyclization of squalene occurs with oxidation, methyl group transfer reactions, saturation of the side chain, and double bond shifting to produce cholesterol.3
Cholesterol is also unique in that, unlike other lipids, it is not readily catabolized by most cells and, therefore, does not serve as a source of fuel. Cholesterol can, however, be converted in the liver to primary bile acids, such as cholic acid (Fig. 15.1) and chenodeoxycholic acid, which promote fat absorption in the intestine by acting as detergents. A small amount of cholesterol can also be converted by some tissue, such as the adrenal gland, testis, and ovary, to steroid hormones, such as glucocorticoids, mineralocorticoids, and estrogens. Finally, a small amount of cholesterol, after first being converted to 7-dehydrocholesterol, can also be transformed to vitamin D3 in the skin by irradiation from sunlight.
GENERAL LIPOPROTEIN STRUCTURE
The prototypical structure of a lipoprotein particle is shown in Figure 15.2. Lipoproteins are typically spherical in shape and range in size from as small as 10 nm to more than 1 μm (Table 15.1) As the name implies, lipoproteins are composed of both lipids and proteins, called apolipoproteins. The amphipathic cholesterol and phospholipid molecules are primarily found on the surface of lipoproteins as a single monolayer, whereas the hydrophobic and neutral triglyceride and cholesteryl ester molecules are found in the central or core region and thus are micelles (Fig. 15.2). Because the main role of lipoproteins is the delivery of fuel to peripheral cells, the core of the lipoprotein particle essentially represents the cargo that is being transported by lipoproteins. The size of the lipoprotein particle correlates with its core neutral lipid content. The larger lipoprotein particles have correspondingly larger core regions and, therefore, contain relatively more triglyceride and cholesteryl ester. The larger lipoprotein particles also contain more lipid relative to protein and thus are lighter in density. The various lipoprotein particles were originally separated by ultracentrifugation into different density fractions (chylomicrons [chylos], VLDL, LDL, and HDL), which still form the basis for the most commonly used lipoprotein classification system (Table 15.1).
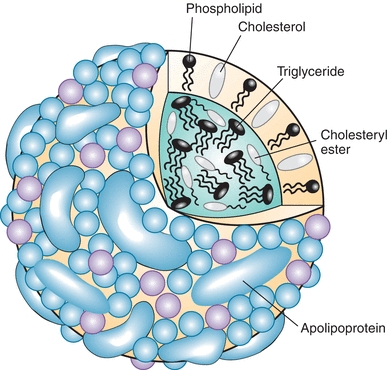
FIGURE 15.2 Model of lipoprotein structure.
TABLE 15.1 Characteristics of the Major Human Lipoproteins
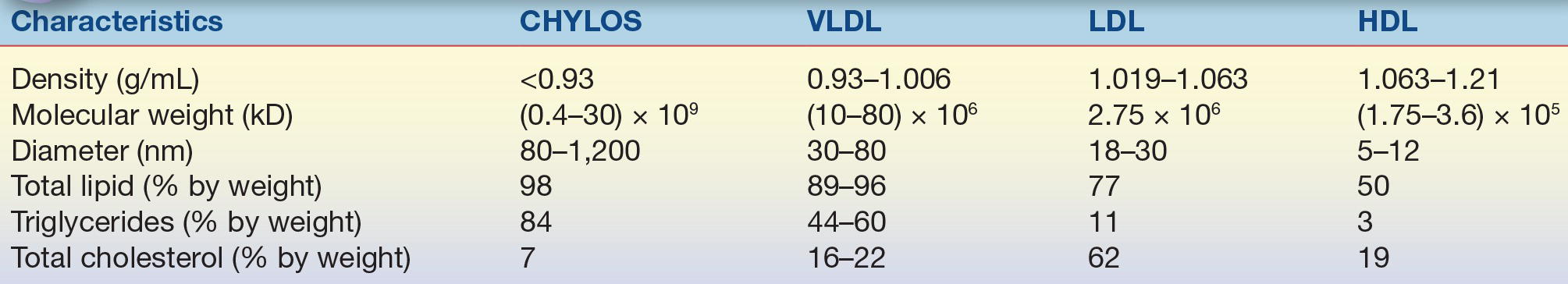
CHYLOS, chylomicrons; VLDL, very-low-density lipoprotein; LDL, low-density lipoprotein; HDL, high-density lipoprotein.
Apolipoproteins are primarily located on the surface of lipoprotein particles (Table 15.2). They help maintain the structural integrity of lipoproteins and also serve as ligands for cell receptors and as activators and inhibitors of the various enzymes that modify lipoprotein particles (Table 15.2). Apolipoproteins contain a structural motif called an amphipathic alpha helix, which accounts for the ability of these proteins to bind to lipids. Amphipathic helices are protein segments arranged in coils so that the hydrophobic amino acid residues interact with lipids, whereas the part of the helix containing hydrophilic amino acids faces away from the lipids and toward the aqueous environment.
TABLE 15.2 Characteristics of the Major Human Apolipoproteins
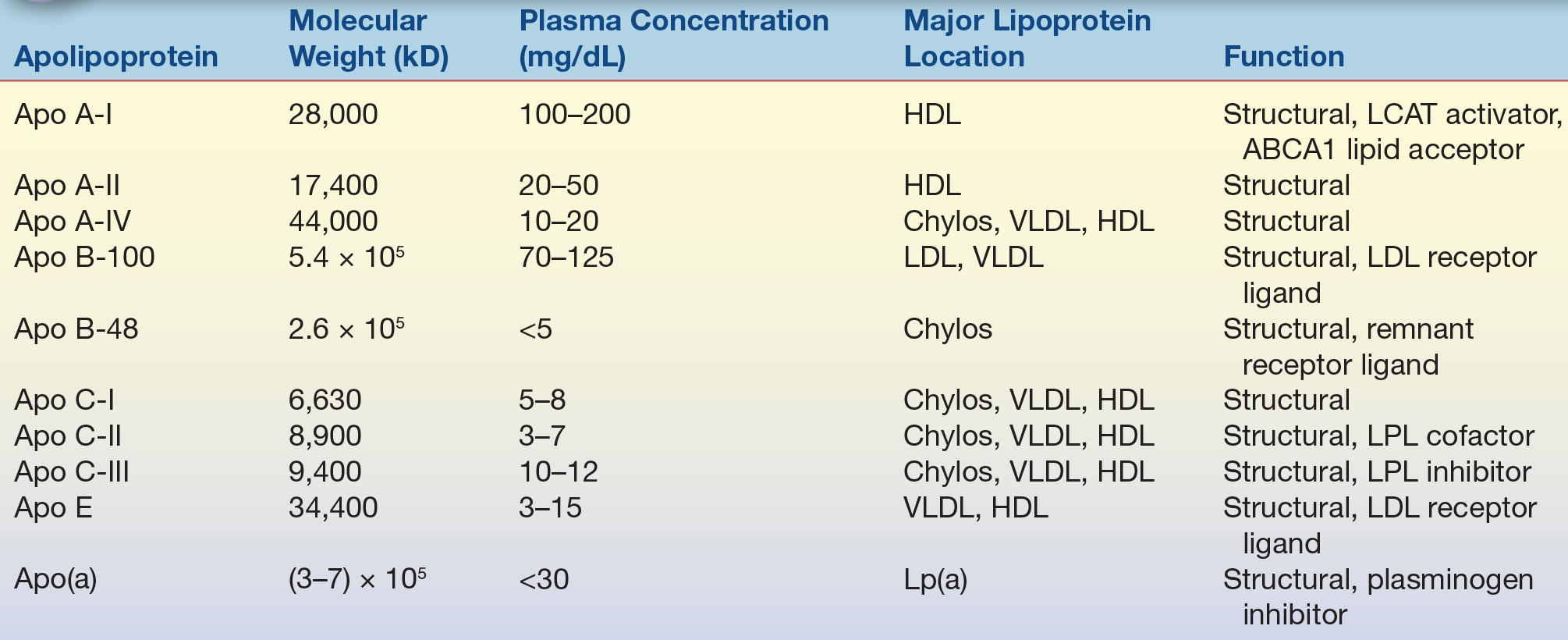
HDL, high-density lipoprotein; Chylos, chylomicrons; LCAT, lecithin–cholesterol acyltransferase; VLDL, very-low-density lipoprotein; LDL, low-density lipoprotein; LPL, lipoprotein lipase.
Apolipoprotein (apo) A-I, the major protein of HDL, is frequently used as a measure of the amount of the antiatherogenic HDL present in plasma.4 Apo B is a large protein with a molecular weight of approximately 500 kD and the principal protein of LDL, VLDL, and chylomicrons.5 Apo B exists in two forms: apo B-100 and apo B-48. Apo B-100 is found in LDL and VLDL and is a ligand for the LDL receptor,6 and it is, therefore, critical in the uptake of LDL by cells. Apo B-48, exclusively found in chylomicrons, is essentially the first 48% or half of the apo B molecule and is produced by posttranscriptional editing of the apo B-100 mRNA found in chromosome 2.7 Apo B also contains amphipathic alpha helices but unlike other apolipoproteins also contain amphipathic beta sheets that avidly bind to lipids. Apo B-100 can also be found covalently linked to apo (a),8 a plasminogen-like protein that is found in a proatherogenic lipoprotein particle called lipoprotein (a) [Lp(a)]. Apo E, another important apolipoprotein found in many types of lipoproteins (LDL, VLDL, and HDL), also serves as a ligand for the LDL receptor and the chylomicron remnant receptor. There are three major isoforms of apo E: apo E2, E3, and E4. The apo E isoforms affect lipoprotein metabolism because they differ in their ability to interact with the LDL receptor. For example, patients who are homozygous for the apo E2 isoform are at an increased risk for developing type III hyperlipoproteinemia and have low affinity for the LDL receptor. The connection with lipid metabolism is not completely understood, but individuals with the apo E4 isoform have higher affinity for the LDL receptor and have been shown to have an increased risk of developing Alzheimer’s disease.10,11
Chylomicrons
Chylomicrons, which contain apo B-48, are the largest and the least dense of the lipoprotein particles, having diameters as large as 1,200 nm9 (Table 15.1). Because of their large size, they scatter light, which accounts for the turbidity or milky appearance of postprandial plasma. Because they are so light, they also readily float to the top of plasma when stored for hours or overnight at 4°C and form a creamy layer. Chylomicrons are produced by the intestine, where they are packaged with absorbed dietary lipids and apolipoproteins. Once they enter the circulation, triglycerides and cholesteryl esters in chylomicrons are rapidly hydrolyzed by lipases, such as lipoprotein lipase (LPL), and, within a few hours, are transformed into chylomicron remnant particles, which are recognized by proteoglycans and remnant receptors in the liver, facilitating their uptake.9 The principal role of chylomicrons is the delivery of dietary lipids to hepatic and peripheral cells.
Very-Low-Density Lipoproteins
VLDL is produced primarily by the liver and contains apo B-100, the main apolipoprotein, apo E, and apo Cs; like chylomicrons, they are also rich in triglycerides. They are the major carriers of endogenous (hepatic-derived) triglycerides and transfer triglycerides from the liver to peripheral tissue mostly during the fasting state for energy utilization and storage. Like chylomicrons, they also reflect light and account for most of the turbidity observed in fasting hyperlipidemic plasma specimens, although they do not form a creamy top layer like chylomicrons, because they are smaller and less buoyant (Table 15.1). Excess dietary intake of carbohydrate, saturated fatty acids, and trans fatty acids enhances the hepatic synthesis of triglycerides, which in turn increases VLDL production. Free fatty acids released into the circulation by adipocytes are avidly taken up by the liver, which also stimulates VLDL secretion.
Intermediate-Density Lipoproteins
Intermediate-density lipoproteins (IDLs), also referred to as VLDL remnants, normally only exist transiently during the conversion of VLDL to LDL. The triglyceride and cholesterol contents of IDL are intermediate between those of VLDL and LDL. Normally, the conversion of VLDL to IDL proceeds so efficiently that appreciable quantities of IDL usually do not accumulate in the plasma after an overnight fast; thus, IDLs are not typically present in high quantities in normal plasma. In patients with type III hyperlipoproteinemia (dysbetalipoproteinemia or broad beta disease), a rare inborn error of metabolism, elevated levels of IDLs can be found in plasma. This defect is due to an abnormal form of apo E that delays the clearance of IDL. Individuals with this disorder are at a significant risk for peripheral vascular disease (PVD) and coronary artery disease (CAD), presumably because these lipoprotein particles like LDL are damaging to the vessel wall.
Low-Density Lipoproteins
LDL primarily contains apo B-100 and is more cholesterol rich than other apo B–containing lipoproteins (Table 15.1). They form as a consequence of the lipolysis of VLDL. LDL is readily taken up by cells via the LDL receptor in the liver and peripheral cells. In addition, because LDL particles are significantly smaller than VLDL particles and chylomicrons, they can infiltrate into the extracellular space of the vessel wall, where they can be oxidized and taken up by macrophages through various scavenger receptors.12 Macrophages that take up too much lipid become filled with intracellular lipid drops and turn into foam cells, which is the predominant cell type of fatty streaks, an early precursor of atherosclerotic plaques.
LDL particles can exist in various sizes and compositions and have been separated into as many as eight subclasses through density ultracentrifugation or gradient gel electrophoresis.13, 14 15 The LDL subclasses differ largely in their content of core lipids; the smaller particles are denser and have relatively more triglyceride than cholesteryl esters. Recently, there has been great interest in measuring LDL subfractions, because small, dense, LDL particles have been shown to be more proatherogenic and may be a better marker for CHD risk.
Lipoprotein (a)
Lp(a) particles are LDL-like particles that contain one molecule of apo (a) linked to apo B-100 by a single disulfide bond. Lp(a) particles are heterogeneous in both size and density, as a result of a differing number of repeating peptide sequences, called kringles, in the apo (a) portion of the molecule. Lp(a) is larger than LDL and has a higher lipid content and a slightly lower density. The concentration of Lp(a) is inversely related to the size of the isoform; the larger size isoforms are not as efficiently secreted from the liver. Plasma levels of Lp(a) vary widely among individuals in the general population but remain relatively constant within an individual. Lp(a) appears to be poorly cleared by the LDL receptor, but the kidney has been postulated as the site of removal.16
Elevated levels of Lp(a) (>30 mg/dL) are now known to increase the risk of premature CHD and stroke. Because the kringle domains of Lp(a) have a high level of homology with plasminogen, a precursor of plasmin that promotes clot lysis via fibrin cleavage, it has been proposed that Lp(a) may compete with plasminogen for binding sites on endothelium and on fibrin, thereby promoting clotting. Clinical studies have demonstrated increasing risk of both myocardial infarction and stroke with increasing Lp(a) concentration; however, the measurement of Lp(a) is often underutilized in clinical practice. Part of the reason is due to the fact that the accurate measurement of Lp(a) is difficult and specific therapies for reducing its concentration in blood are limited but several are in development. Measuring Lp(a) is recommended for patients with a strong family history of CHD, particularly in the absence of other known risk factors, such as increased LDL-C, or for patients that develop CHD on statin therapy or for patients with premature aortic stenosis, which has been shown to be caused by high Lp(a) levels.
High-Density Lipoproteins
HDL, the smallest and most dense lipoprotein particle, is synthesized by both the liver and the intestine (Table 15.1). HDL can exist as either disk-shaped particles or, more commonly, spherical particles.4 Discoidal HDL typically contains two molecules of apo A-I, which form a ring around a central lipid bilayer of phospholipid and cholesterol. Discoidal HDL is believed to represent nascent or newly secreted HDL and is the most active form in removing excess cholesterol from peripheral cells. The ability of HDL to remove cholesterol from cells, called reverse cholesterol transport, is one of the main mechanisms proposed to explain the antiatherogenic property of HDL. When discoidal HDL has acquired an additional lipid, cholesteryl esters and triglycerides form a core region between its phospholipid bilayer, which transforms discoidal HDL into spherical HDL. HDL is highly heterogeneous in size and lipid and protein composition and is separable into as many as 13 or 14 different subfractions. There are two major types of spherical HDL based on density differences: HDL2 (1.063 to 1.125 g/mL) and HDL3 (1.125 to 1.21 g/mL). HDL2 particles are larger in size, less dense, and richer in lipid than HDL3 and may be more efficient in the delivery of lipids to the liver.
Lipoprotein X
Lipoprotein X is an abnormal lipoprotein present in patients with biliary cirrhosis or cholestasis and in patients with mutations in lecithin–cholesterol acyltransferase (LCAT), the enzyme that esterifies cholesterol. Lipoprotein X is different from other lipoproteins in the endogenous pathway due to the lack of apo B-100. Phospholipids and nonesterified cholesterol are its main lipid components (~90% by weight) and albumin and apo C are the main protein components (<10% by weight). Lipoprotein X is mainly removed by the reticuloendothelial system of the liver and the spleen. Other organs, such as the kidney, also actively clear lipoprotein X from the plasma, which may account for renal disease in patients with genetic LCAT deficiency.17 Lipoprotein X is typically included in the LDL-C value as calculated by the Friedewald equation, as well as in some direct methods for LDL-C but can be separated by LDL-C by ultracentrifugation.
LIPOPROTEIN PHYSIOLOGY AND METABOLISM
The four major pathways involved in lipoprotein metabolism are shown in Figure 15.3. The lipid absorption pathway, the exogenous pathway, and the endogenous pathway all depend on apo B–containing lipoprotein particles and can be viewed as the process to transport dietary lipid and hepatic-derived lipid to peripheral cells. In terms of energy metabolism, these three pathways are critical in the transport to peripheral cells of fatty acids, which are generated during the lipolysis of triglycerides and, to a lesser degree, cholesteryl esters on lipoproteins. In regard to the pathogenesis of atherosclerosis, the net result of these three pathways is the net delivery or forward transport of cholesterol to peripheral cells, which can lead to atherosclerosis when the cells in the vessel wall accumulate too much cholesterol. Peripheral cells are prone to accumulating cholesterol because they also synthesize their own cholesterol, and, unlike liver cells, they do not have the enzymatic pathways to catabolize cholesterol. Furthermore, cholesterol is relatively water insoluble and cannot readily diffuse away from its site of deposition or synthesis.
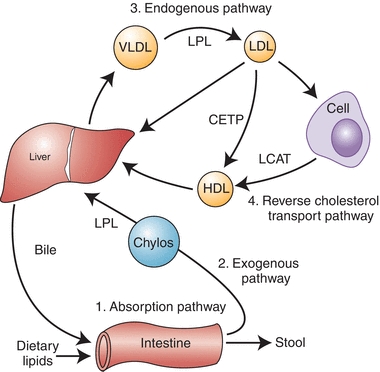
FIGURE 15.3 Diagram of major lipoprotein metabolism pathways.
The principal way that peripheral cells maintain their cholesterol equilibrium is the reverse cholesterol transport pathway (Fig. 15.3), which is mediated by HDL. In this pathway, excess cholesterol from peripheral cells is transported back to the liver, where it can be excreted into the bile as free cholesterol or after being converted to bile acids. The liver is, therefore, involved in both forward and reverse cholesterol transport pathways and, in many ways, acts as a buffer in helping the body maintain its overall cholesterol homeostasis. There are several genetic defects in the genes that encode for proteins in the forward and reverse cholesterol transport pathways, which result in a predisposition for atherosclerosis. The majority of individuals with CAD, however, do not have a clear, single, genetic defect but instead have multiple genetic variations or gene polymorphisms that most likely interact with various lifestyle habits, such as exercise, diet, and smoking, leading to a predisposition for disease.
Lipid Absorption
Because fats are water insoluble, special mechanisms are required to facilitate the intestinal absorption of the 60 to 130 g of fat consumed per day in a typical Western diet. During the process of digestion, pancreatic lipase, by cleaving off fatty acids, first converts dietary lipids into more polar compounds with amphipathic properties. Thus, triglycerides are transformed into monoglycerides and diglycerides; cholesterol esters are transformed into free cholesterol; and phospholipids are transformed into lysophospholipids. These amphipathic lipids in the intestinal lumen form large aggregates with bile acids called micelles. Lipid absorption occurs when the micelles come in contact with the microvillus membranes of the intestinal mucosal cells. Absorption of some of these lipids may occur via a passive transfer process; however, recent evidence suggests that, in some cases, it might also be facilitated by specific transporters, such as the NPC1L-1 transporter for cholesterol. Short-chain free fatty acids, with 10 or fewer carbon atoms, can readily pass directly into the portal circulation and are carried by albumin to the liver. The absorbed long-chain fatty acids, monoglycerides, and diglycerides are reesterified in intestinal cells to form triglycerides and cholesteryl esters. The newly formed triglycerides and cholesteryl esters are then packaged into chylomicrons by the microsomal transfer protein, along with apo B-48.
Triglyceride absorption is efficient; greater than 90% of dietary triglycerides are taken up by the intestine. In contrast, only about half of the 500 mg of cholesterol in the typical diet is absorbed each day. Even a smaller fraction of plant sterols is absorbed by the intestine. Recently, a specific transport system, involving the ABCG5 and ABCG8 transporters, has been described that prevents excess absorption of dietary cholesterol and plant sterols. Individuals with defective ABCG5 or ABCG8 transporters have a disease called sitosterolemia and have a predisposition for atherosclerosis and xanthomatosis because of increased cholesterol and plant sterol absorption.18 ABCG5 and ABCG8 are also present in the liver and defects in these proteins also impair the elimination of plant sterols into bile for removal from the body.19
Exogenous Pathway
The newly synthesized chylomicrons in the intestine (Fig. 15.3) are initially secreted into the lacteals and then pass into the lymphatic ducts and eventually enter the circulation by way of the thoracic duct. After entering the circulation, chylomicrons interact with proteoglycans, such as heparin sulfate, on the luminal surface of capillaries in various tissues, such as skeletal muscle, heart, and adipose tissue. The proteoglycans along with a specific protein called GPIHBPI on capillaries also promote the binding of LPL,20 which hydrolyzes triglycerides on chylomicrons. The free fatty acids and glycerol generated by the hydrolysis of triglycerides by LPL can then be taken up by cells and used as a source of energy. Excess fatty acids, particularly in fat cells (adipocytes), are reesterified into triglycerides for long-term energy storage in intracellular lipid drops. A key protein in triglyceride metabolism is apo C-II, which is found on VLDL and is critical in the activation of LPL. Hormone-sensitive lipase, another lipase that is found inside adipose cells, releases free fatty acids from triglycerides in stored fat, during fasting, when energy sources from carbohydrates are insufficient for the body’s energy needs. The hormones epinephrine and cortisol play a key role in the mobilization and hydrolysis of triglycerides from adipocytes, whereas insulin right after a meal prevents lipolysis by adipocytes and promotes fat storage and glucose utilization.
During lipolysis of chylomicrons, there is a transfer of lipid (mainly triglyceride) and apolipoproteins (apo A-I and C-II) onto HDL, and chylomicrons are converted within a few hours after a meal into chylomicron remnant particles. Chylomicron remnants are rapidly taken up by the liver through interaction of apo E with the LDL receptor and another receptor on the surface of liver cells called LDL-related receptor protein.21 Once in the liver, lysosomal enzymes break down the remnant particles to release free fatty acids, free cholesterol, and amino acids. Some cholesterol is converted to bile acids. Both bile acids and free cholesterol are directly excreted into the bile, but not all of the excreted cholesterol and bile salt exit the body. As previously described, approximately half of the excreted biliary cholesterol is reabsorbed by the intestine, with the remainder appearing in the stool, as fecal neutral steroids. In the case of bile acids, almost all of the bile acids are reabsorbed and reused by the liver for bile production.
Endogenous Pathway
Most triglycerides in the liver that are packaged into VLDL are derived from the diet after recirculation from adipose tissue. Normally, only a small fraction is synthesized de novo in the liver from dietary carbohydrate. VLDL particles, once secreted into the circulation, undergo a lipolytic process similar to that of chylomicrons (Fig. 15.3). VLDL loses core lipids, causing dissociation and transfer of apolipoproteins and phospholipids to other lipoprotein particles like HDL, primarily by the action of LPL, resulting in the conversion of VLDL to denser particles, called IDLs or VLDL remnants. IDL persists for short periods of time and receives cholesterol esters from HDL in exchange for triglycerides via cholesteryl ester transport protein. As with chylomicron remnants, IDL is taken up by the liver via apo E and the LDL receptor and the triglyceride in IDL are removed by hepatic triglyceride lipase, located on hepatic endothelial cells, producing LDL. About half of VLDL is eventually completely converted to LDL, and the remainder is taken up as VLDL remnants by the liver remnant receptors.
LDL particles are the major lipoproteins responsible for the delivery of exogenous cholesterol to peripheral cells due to the efficient uptake of LDL by the LDL receptors.16 LDL can pass between capillary endothelial cells and bind to LDL receptors on cell membranes that recognize apo B-100. Once bound to LDL receptors, they are endocytosed by cells and transported to the lysosome, where they are degraded. The triglycerides in LDL are converted by acid lipase into free fatty acids and glycerol and further metabolized by the cell for energy or are reesterified and stored in lipid drops for later use. Free cholesterol derived from degraded LDL can be used for membrane biosynthesis, and excess cholesterol is converted by acyl-CoA–cholesterol acyltransferase (ACAT) into cholesteryl esters and stored in intracellular lipid drops. The regulation of cellular cholesterol biosynthesis is, in part, coordinated by the availability of cholesterol delivered by the LDL receptor. Many enzymes in the cholesterol biosynthetic pathway (e.g., HMG-CoA reductase, the main target for the cholesterol-lowering statin-type drugs) are down-regulated, along with the LDL receptor, when there is excess cellular cholesterol by a complex mechanism involving both gene regulation and posttranscriptional gene regulation.
Abnormalities in LDL receptor function result in elevation of LDL in the circulation and lead to hypercholesterolemia and premature atherosclerosis. Patients who are heterozygous for a disease called familial hypercholesterolemia (FH), with an incidence of approximately 1:500, have only approximately half the normal LDL receptors, which results in decreased hepatic uptake of LDL by the liver and increased hepatic cholesterol biosynthesis. The LDL that accumulates in the plasma of these individuals often leads to the development of CHD by midadulthood in heterozygotes and even earlier for homozygotes.
Individuals can be classified into two distinct phenotypes based on LDL, namely, types A and B. Type A individuals have large buoyant LDL particles, whereas in type B, LDL particles are smaller.22 Elevated VLDL is associated with an exchange of VLDL triglycerides for cholesterol esters in HDL, resulting in triglyceride-rich HDL with decreased cholesterol content.22 Cholesterol esters in LDL are also exchanged for VLDL triglycerides.22 The triglycerides in LDL and HDL are subsequently removed by the action of lipases, producing smaller, more dense cholesterol-containing particles.22 Small, dense LDL particles have been found to be an independent predictor of CHD and is also associated with increased risk of metabolic syndrome, which is a group of interrelated metabolic risk factors that appear to promote the development of atherosclerotic cardiovascular disease.22 Small, dense LDL particles have a longer half-life in circulation than normal LDL and are more likely to be modified and taken up by scavenger receptors on macrophage, which will develop into foam cells, one of the critical steps in the development of atheroma.22 It is difficult, however, to define the degree of increased risk with small, dense LDL particles quantitatively because of the common association between small LDLs with other risk factors of atherogenic dyslipidemia.
Reverse Cholesterol Transport Pathway
As previously described, one of the major roles of HDL is to maintain the equilibrium of cholesterol in peripheral cells by the reverse cholesterol transport pathway (Fig. 15.3). HDL is believed to remove excess cholesterol from cells by multiple pathways. In the aqueous diffusion pathway, HDL acts as a sink for the small amount of cholesterol that can diffuse away from the cells. Although cholesterol is relatively water insoluble, because it is an amphipathic lipid, it is soluble in plasma in micromolar amounts and can spontaneously dissociate from the surface of cell membranes and enter the extracellular fluid. Some free cholesterol will then bind to nascent HDL in the extracellular space, and once bound, it becomes trapped in lipoproteins after it is converted to cholesteryl ester by the enzyme LCAT, which resides on nascent HDL and is activated by its cofactor, apo A-I. The nascent HDL is first converted to HDL3. The formation of cholesteryl ester via LCAT increases the capacity of the surface of HDL3 particle to absorb more free cholesterol from cell membranes along with apo C-I, C-II, C-III, and E and phospholipids from VLDL and chylomicrons and is converted to HDL2. HDL2 can then directly deliver cholesterol to the liver by the scavenger receptor type B1 (SR-B1) and, possibly, other receptors. HDL particles can also transfer cholesteryl esters to chylomicrons and VLDL remnants to be transported to liver. Approximately half of the cholesterol on HDL is returned to the liver by the LDL receptor, after first being transferred from HDL to LDL by the cholesteryl ester transfer protein (CETP), which connects the forward and reverse cholesterol transport pathways (Fig. 15.3). Cholesterol that reaches the liver is then directly excreted into the bile or first converted to a bile acid before excretion.
Another pathway in which HDL mediates the removal of cholesterol from cells involves the ABCA1 transporter. The ABCA1 transporter is a member of the ATP-binding cassette transporter family,23 which pumps various ligands across the plasma membrane. Defects in the gene for the ABCA1 transporter lead to Tangier disease, a disorder associated with low HDL and a predisposition to premature CHD.24 The exact substrate for the ABCA1 transporter is not known, but it is believed that the transporter modifies the plasma membrane by transferring a lipid, which then enables apo A-I that has dissociated from HDL to bind to the cell membrane. In a detergent-like extraction mechanism, apo A-I then removes excess cholesterol and phospholipid from the plasma membrane of cells to form a discoidal-shaped HDL particle. The newly formed HDL is then competent to accept additional cholesterol by the aqueous diffusion pathway and is eventually converted into spherical HDL by the action of LCAT (Fig. 15.3). Recently, ABCG1, ABCG5 and ABCG8, which are other ABC transporters, have been described to facilitate the efflux of cholesterol to lipid-rich spherical HDL via a mechanism that appears to be different than the ABCA1 transporter.25
The ability of the body to remove excess cholesterol has for decades been measured in vitro using a cell-based assay, which tests the capacity of cholesterol-loaded macrophages to efflux cholesterol through the ABCA1 transporter.26 This test provides additional knowledge about the function of the HDL particle beyond the HDL-C measurement. Recently, it was reported that efflux capacity, independent of HDL-C levels, was inversely correlated with intima–media thickness and the presence of CHD.27 In fact, efflux capacity was a better predictor of disease status than conventional risk factors such as diabetes, smoking, LDL-C, and hypertension.27 The importance of the efflux capacity was also reported in a population-based study where low levels were associated with incident atherosclerotic cardiovascular disease suggesting an impaired efflux capacity in the investigated subjects.28
LIPID AND LIPOPROTEIN POPULATION DISTRIBUTIONS
Serum lipoprotein concentrations differ between adult men and women, primarily as a result of differences in sex hormone levels, with women having, on average, higher HDL-C levels and lower total cholesterol and triglyceride levels than men. The difference in total cholesterol, however, disappears after menopause as estrogen decreases. Men and women both show a tendency toward increased total cholesterol, LDL-C, and triglyceride concentrations with age. HDL-C concentrations generally remain stable after the onset of puberty and do not drop in women with the onset of menopause. General adult reference ranges are shown in Table 15.3.
TABLE 15.3 Adult Reference Ranges For Lipids

HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol.
Circulating levels of total cholesterol, LDL-C, and triglycerides in young children are generally much lower than those seen in adults.29 In addition, concentrations do not significantly differ between boys and girls. HDL-C levels for both boys and girls are comparable to those of adult women. At the onset of puberty, however, HDL-C concentrations in boys fall to adult male levels, a drop of approximately 20%, whereas those of girls do not significantly change. It is the lower concentration of HDL-C in men, combined with their higher LDL-C and triglyceride levels, which accounts for much of the increased risk of premature heart disease in men.
The incidence of heart disease is strongly associated with serum cholesterol concentration, particularly LDL-C. Comparisons across different societies show that eating less animal fat and more grains, fruits, and vegetables, as is common in many Asian populations, is associated with lower LDL-C and lower rates of heart disease compared with societies that ingest more fat, particularly animal fat, and are more sedentary. These differences can be attributed to both genetic and lifestyle factors in the various countries and ethnic groups. The importance of diet was clearly shown in a study that compared the dietary patterns and heart disease rates in Japanese men living in Japan, Hawaii, and California.30 In this study, as dietary intake became more Westernized, with increased consumption of fat and cholesterol, the LDL-C concentrations increased, as did the rates of heart disease. Japanese men living in California were found to have much higher rates of heart disease than Japanese men living in Japan; those in Hawaii were intermediate. Within societies in which diet tends to be more homogeneous, LDL-C levels become somewhat less discriminatory as a risk factor, and HDL-C levels become more important as a negative risk factor.
CASE STUDY 15.1
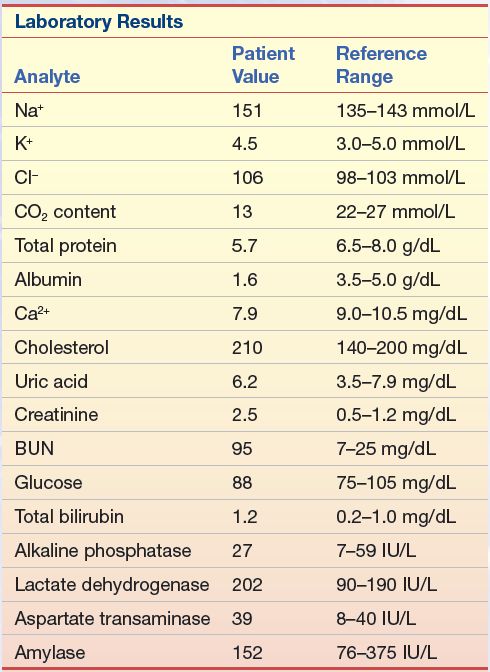
A 52-year-old man went to his physician for a physical examination. The patient had been a district manager for an automobile insurance company for the past 10 years and was a 24-lb overweight. He had missed his last two appointments with the physician because of business. The urinalysis dipstick finding was not remarkable. His blood pressure was elevated. The blood chemistry results are listed in Case Study Table 15.1.1.
Questions
1. Given the abnormal tests, what additional information would you like to have?
2. If this patient had triglycerides of 100 mg/dL (1.1 mmol/L) and an HDL-C of 23 mg/dL (0.6 mmol/L), what would be his calculated LDL-C value?
3. If, however, his triglycerides were 476 mg/dL (5.4 mmol/L), with an HDL-C of 23 mg/dL (0.6 mmol/L), what would be his calculated LDL-C value?
The NCEP was formed to alert the American population to the risk factors associated with heart disease. The NCEP has used panels of experts, including Adult Treatment Panels (ATPs), the Children and Adolescents Treatment Panel, and the Laboratory Standardization Panel, to produce various recommendations.31 In 1988, the first NCEP ATP developed a list of heart disease risk factors. These guidelines were most recently updated by ATP IV in 2013.32
In general, the ATP guidelines and other similar guidelines are based on the central tenant that lifestyle changes, such as a low-fat diet, increased physical activity, and weight control, should be the first-line treatment for all patients at risk for cardiovascular disease (CVD) but that more aggressive drug therapy should also be considered for the highest risk individuals and/or for those that do not respond to lifestyle changes. According to the new ATP IV guidelines,32,33 patients at high risk for CVD (diabetics and preexisting CVD) or those with an LDL-C greater than 190 mg/dL are advised to initiate simultaneous lifestyle changes along with drug therapy, defined as high-dose statin in order to reduce LDL-C by 50% from baseline.32,33 Lower-risk patients between age 40 and 75 should have their 10-year CVD risk calculated and be treated with high- or moderate-dose statin if their risk score is greater than 7.5% and moderate-dose statin when their risk score is between 5% and 7.5%. For those patients with a 10-year risk less than 5% or if they fall outside the age range for the risk calculator (<40 years or >75 years), only lifestyle changes would typically be recommended, but statins could be considered if they have other extenuating circumstances, such as a strong family history or at risk based on other risk marker like CRP. Repeat laboratory testing is recommended to monitor compliance and to determine if at least a 50% reduction in LDL-C is achieved, but there are no longer any specific treatment target goals for LDL-C like in past guidelines. The current list of risk factors used in calculating the 10-year risk of CVD is shown in Table 15.4. It is recommended that all adults (20 years and older) have a fasting lipoprotein profile performed (total cholesterol, LDL-C, and HDL-C and triglycerides) once every 5 years. As described below, the Children and Adolescents Treatment Panel has developed similar guidelines for CVD risk assessment in the pediatric population.29
TABLE 15.4 Coronary Heart Disease Risk Factors Determined by the NCEP Adult Treatment Panels

CHD, coronary heart disease; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol.
Dyslipidemia and Children
It is well recognized and documented that the atherosclerotic process begins in childhood, with dyslipidemia as one of the main risk factors for cardiovascular disease (CVD). A recent analysis of National Health and Nutrition Examination Survey data collected between 1998 and 2010 estimated that 24.6% of US children aged 9 to 11 had either abnormally low HDL cholesterol or elevated non–HDL cholesterol.34 Elevated total cholesterol, LDL-C, VLDL-C, and decreased HDL-C concentrations are significantly associated with early atherosclerotic lesions in adolescent and young adults.35 Thus, early identification and treatment of children with hyperlipidemia is likely to delay the atherosclerotic process and reduce CVD risk in adulthood. Guidelines for the assessment and treatment of CVD risk in children and adolescents have been published by the National Heart, Lung, and Blood Institute (NHLBI).29
National Cholesterol Education Program
The goal of the NCEP (created in 1985 by NHLBI) is to educate the public and medical professionals about the benefits of lowering cholesterol levels to reduce the risk of CHD. The NCEP published the first set of pediatric guidelines for dyslipidemia in 1992.29 These guidelines had two approaches: both diet and lifestyle changes for all children and a targeted lipid screening for individual children with a family history of heart disease, FH, or familial combined hyperlipidemia (FCH).29 Some considered the reliance of these guidelines on family history to be too restrictive, and several studies demonstrated a significant number of children with elevated cholesterol levels would have been missed by these guidelines. For example, the Coronary Artery Risk Detection in Appalachian Communities (CARDIAC) Project, which utilized universal screening, found that 36% of children with severe dyslipidemia would have been overlooked.36
National Heart, Lung, and Blood Institute
With childhood obesity on the rise and almost half of obese children having at least one abnormal lipid level, the NHLBI Expert panel in 2011 published a comprehensive updated evidence-based guideline for the identification, management, and treatment of all major CV risk factors (including diabetes, hypertension, cigarette smoking, and obesity) in pediatric patients.29 Universal lipid screening of all children from 9 to 11 years and again at 17 to 21 years with a nonfasting, non–HDL-C levels and targeted lipid screening are both supported by the NHLBI guidelines and endorsed by the American Academy of Pediatrics.29
The 9 to 11 age range is important because that is when then atherosclerotic process appears to accelerate but before the decline in LDL-C seen with puberty. Universal screening is not recommended for children before 9 years of age due to insufficient information on the safety and impact of lipid-lowering medications.29 The NCEP guidelines also recommend selective screening to identify children at high dyslipidemia risk with two fasting lipid profiles between 2 and 8 years and again between 12 and 16 years. NHLBI guidelines provide algorithms to assist clinicians with the screening process, risk factor management, and treatment of dyslipidemia.29 The NHLBI universal screening recommendations for lipid abnormalities in children and adolescents are controversial, and some health care providers have resisted.37 Acceptable lipid levels for nonfasting blood specimens for youth and young adults are shown in Table 15.5.29 If a nonfasting lipid profile reveals a non–HDL-C greater than 145 mg/dL, then two separate fasting lipid profiles should be done at least 2 weeks apart but within 3 months of each other and the results averaged to confirm the abnormality. If an initial fasting lipid profile reveals an LDL-C ≥130 mg/dL, then another fasting lipid profile should be obtained at least 2 weeks later but within 3 months and the results averaged. The NHLBI recommends that children identified with dyslipidemia should begin with diet and lifestyle changes along with patient education and progress to more stringent diet and lifestyle changes.29 If dyslipidemia persists, then pharmacological treatment and/or referral to a lipid specialist may be recommended for children aged ≥10 years.29 Likewise, children with a genetic dyslipidemia, such as FH, should also be referred to a lipid specialist for more aggressive-type therapy.29 Future studies are needed, however, to firmly establish if treatment of hyperlipidemia early in life prevents CHD in adulthood.
TABLE 15.5 Lipid Cut Points (mg/dL)
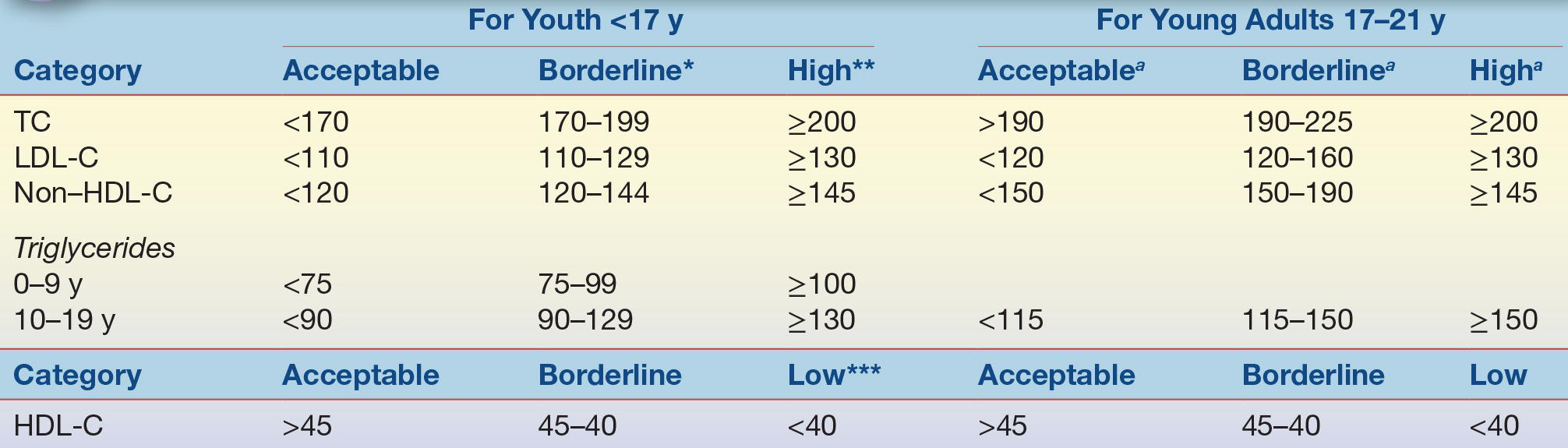
Values represent approximately the *75th and **95th percentiles and approximately the ***10th percentile for low HDL-C.
aThe cut points for TC, HDL-C, and non–HDL-C represent the 95th percentile for 20- to 24-year-old subjects; acceptable values are at the <75th percentile. For HDL-C, low is <25th percentile and acceptable values are >50th percentile.
HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; TC, total cholesterol. From Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents, National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011;128(suppl 5):S213–S256.
The NCEP Laboratory Standardization Panel and its successor, the Lipoprotein Measurement Working Group,38, 39, 40 set laboratory guidelines for acceptable precision and accuracy when measuring total cholesterol, triglycerides, and lipoprotein cholesterol (HDL-C and LDL-C) (Table 15.6).
TABLE 15.6 NCEP Analytic Performance Goals
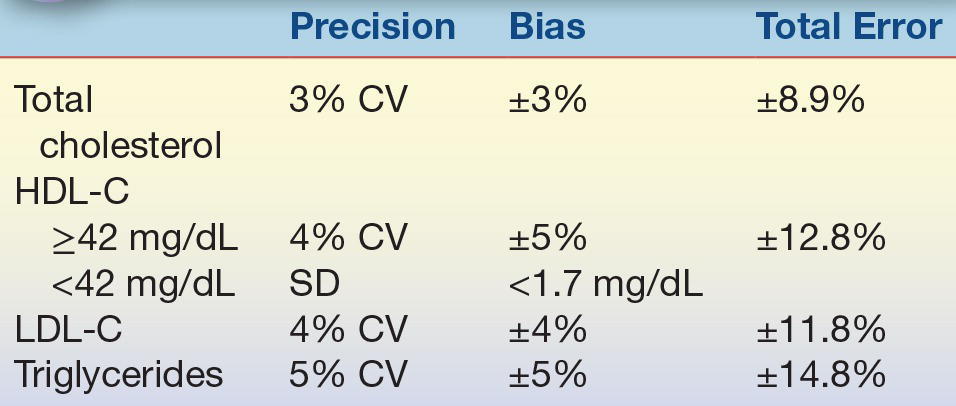
CV, coefficient of variation; HDL-C, high-density lipoprotein cholesterol; SD, standard deviation; LDL-C, low-density lipoprotein cholesterol.
Obviously, the best way to reduce the prevalence of heart disease is through prevention. Learning and practicing good dietary and exercise patterns early in life, maintaining these patterns throughout life,41 refraining from smoking, and controlling blood pressure are important means for reducing the incidence of CHD and stroke. Lipoprotein profile measurements provide a method of identifying individuals who may have levels that put them at risk so that they can receive appropriate treatment. Treatment of other diseases that may affect lipoproteins, such as diabetes mellitus, hypothyroidism, and renal disease, is also important. A prudent diet, low in fat, with a caloric intake adjusted to meet and maintain ideal body weight, along with regular exercise, can significantly reduce the risk of heart disease, stroke, diabetes, and cancer.42 Additionally, saturated fat is more atherogenic than unsaturated fat.41 The American Heart Association has recommended dietary guidelines for the intake of fat and cholesterol for most adult Americans (Table 15.7).
TABLE 15.7 Composition of the Therapeutic Lifestyle Changes Diet Recommended by the NCEP Adult Treatment Panel III (Compared with the Average American Diet)
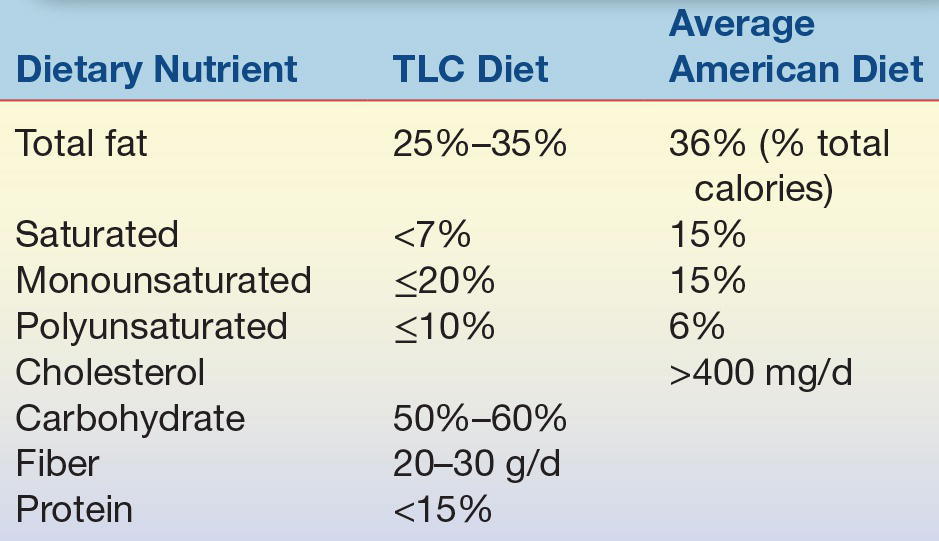
TLC, therapeutic lifestyle changes.
DIAGNOSIS AND TREATMENT OF LIPID DISORDERS
Diseases associated with abnormal lipid concentrations are referred to as dyslipidemias. They can be caused directly by genetic abnormalities or through environmental/lifestyle imbalances, or they can develop secondarily, as a consequence of other diseases. Dyslipidemias are generally defined by the clinical characteristics of patients and the results of laboratory tests and are not necessarily defined by the specific genetic defect associated with the abnormality. Many, but not all, dyslipidemias, regardless of etiology, are associated with CHD or arteriosclerosis.
Arteriosclerosis
In the United States and many other developed countries, arteriosclerosis is the single leading cause of death and disability. The mortality rate has leveled off and started to decrease in the United States in the past three decades, partly as a result of advances in diagnosis and treatment but also because of changes in lifestyle in the American population. This increased awareness of the importance of diet and exercise in preventing CHD has resulted in an overall decrease in the average serum cholesterol concentration and in a lower prevalence of heart disease; however, it still exceeds all other causes of death combined. Although as many women as men now develop arteriosclerosis, they typically develop it 10 years later than men, and unfortunately, it sometimes goes unrecognized because of lack of awareness and because symptoms of myocardial infarction are sometimes subtler in women.
The relationship between heart disease and dyslipidemias stems from the deposition of lipids, mainly in the form of esterified cholesterol, in artery walls. This lipid deposition first results in fatty streaks, which are thin streaks of excess fat in macrophages in the subendothelial space. Autopsy studies have shown that fatty streaks occur in almost everyone older than age 15.43 Fatty streaks can develop over time into plaques that contain increased number of smooth muscle cells, extracellular lipid, calcification, and fibrous tissue, which can partially block or occlude blood flow. Also, established plaque for unknown reasons can become vulnerable to rupture or erosion, triggering a thrombosis that can block circulation. When plaque develops in arteries of the arms or legs, it is called PVD; when it develops in the heart, it is referred to as CAD; and, when it develops in the vessels of the brain, it is called cerebrovascular disease. CAD is associated with angina and myocardial infarction, and CVD is associated with stroke. Many genetic and acquired dyslipidemias may also lead to lipid deposits in the liver and kidney, resulting in impaired function of these vital organs. Lipid deposits in skin form nodules called xanthomas, which are often a clue to the presence of an underlying genetic abnormality.
Plaque formation involves repeated cycles of cell injury, followed by infiltration and cell proliferation to repair the site. LDL is believed to play a central role in initiating and promoting plaque formation. It is deposited into the subendothelial space where it is taken up by various cells, including macrophages. This alters the gene and protein expression pattern of these cells and can promote an inflammatory response, particularly when LDL becomes oxidized.44 Injury signals from the evolving plaque trigger the expression of adhesion proteins on endothelial cells and the production of soluble chemotactic proteins from resident macrophages, which promotes the attachment and infiltration of additional macrophages, lymphocytes, and platelets to the plaque. Continual injury and repair lead to additional narrowing of the vessel opening, or lumen, causing the blood to circulate in a nonlaminar manner under greater and greater pressure, which further aggravates plaque formation. The final event leading to complete occlusion of blood flow occurs when there is a hemorrhage into the plaque, which results in the formation of a thrombus that blocks blood flow and precipitates a myocardial infarction.
CASE STUDY 15.2
A 30-year-old man with chest pain was brought to the emergency department after a softball game. He was placed in the coronary care unit when his ECG showed erratic waves in the ST region. A family history revealed that his father died of a heart attack at the age of 45 years. The patient had always been athletic in high school and college, so he had not concerned himself with a routine physical. The laboratory tests listed in Case Study Table 15.2.1 were run.
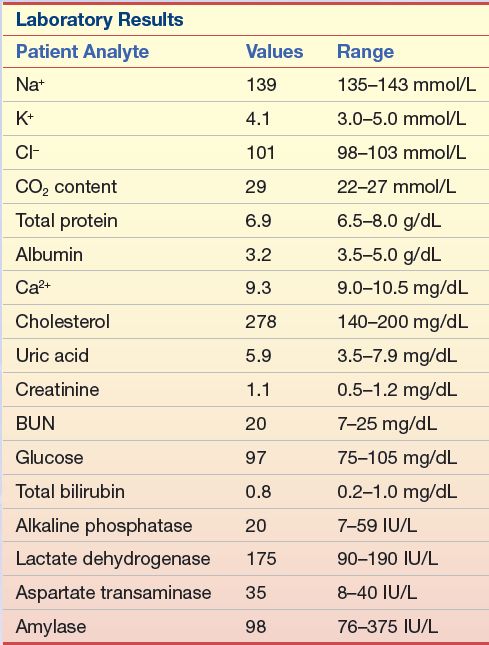
Questions
1. Given the symptoms and the family history, what additional tests should be recommended?
2. If his follow-up total cholesterol remains in the same range after he is released from the hospital, and his triglycerides and HDL-C are within the normal range, what course of treatment should be recommended?
Because lipid deposits in the vessel walls are frequently associated with increased serum concentrations of LDL-C, lowering LDL is an important step in preventing and treating CHD.45, 46, 47 It is estimated that for every 1% decrease in LDL-C concentration, there is a 2% decrease in the risk of developing arteriosclerosis.48 For patients with established heart disease, studies have shown that aggressive treatment to reduce LDL-C levels below 100 mg/dL (2.6 mmol/L) or even lower is effective in the stabilization and sometimes regression of plaques.49 Stabilization of plaque is thought to be at least as important as plaque regression in terms of rupture potential.
In some individuals, high levels of blood cholesterol or triglycerides are caused by genetic abnormalities in which either too much is synthesized or too little is removed. High levels of cholesterol and/or triglycerides in most people, however, are a result of increased consumption of foods rich in fat and cholesterol, smoking, and lack of exercise or a result of other disorders or disease states that affect lipid metabolism, such as diabetes, hypertension, hypothyroidism, obesity, liver and kidney diseases, and alcoholism. Low levels of HDL-C are also associated with increased risk of heart disease, but there are currently limited ways to pharmacologically raise HDL-C levels and it is unclear to what degree low HDL is causal in the pathogenesis of atherosclerosis. Existing drugs for increasing HDL-C are primarily fibric acid derivatives (fibrates) and niacin-containing compounds, but recent studies have failed to show a clear benefit from raising HDL with these drugs.50 Newer drugs that raise HDL-C by inhibiting CETP may play a role in the future, but so far, these drugs have also failed to show a benefit. Other drugs using reconstituted HDL particles or administration of apo A-I mimetic peptides is also being actively investigated.51
Laboratory analyses are an important adjunct to managing patients with dyslipidemia, because accurate measurement of total cholesterol, HDL-C, and LDL-C and triglyceride levels is needed to determine the most appropriate diet or diet and drug therapy. As shown in Table 15.8 from the older ATP III guidelines, individuals on a low-fat diet, who continue to have LDL-C levels of 190 mg/dL (4.9 mmol/L) or higher on repeated measurement, will likely benefit from drug intervention. If they have two or more CAD risk factors and continue to have LDL-C levels of 160 mg/dL (4.1 mmol/L) or higher, they also would benefit from drug therapy. And if they have already been previously diagnosed with heart disease, drug therapy should be considered when the LDL-C level is 130 mg/dL (3.4 mmol/L) or higher. The average of at least two measurements, taken 1 to 8 weeks apart, should be used to determine the best treatment approach.31
TABLE 15.8 Treatment Guidelines Established by the NCEP Adult Treatment Panels (Initial Testing Should Consist of Fasting for ≥12 Hours)
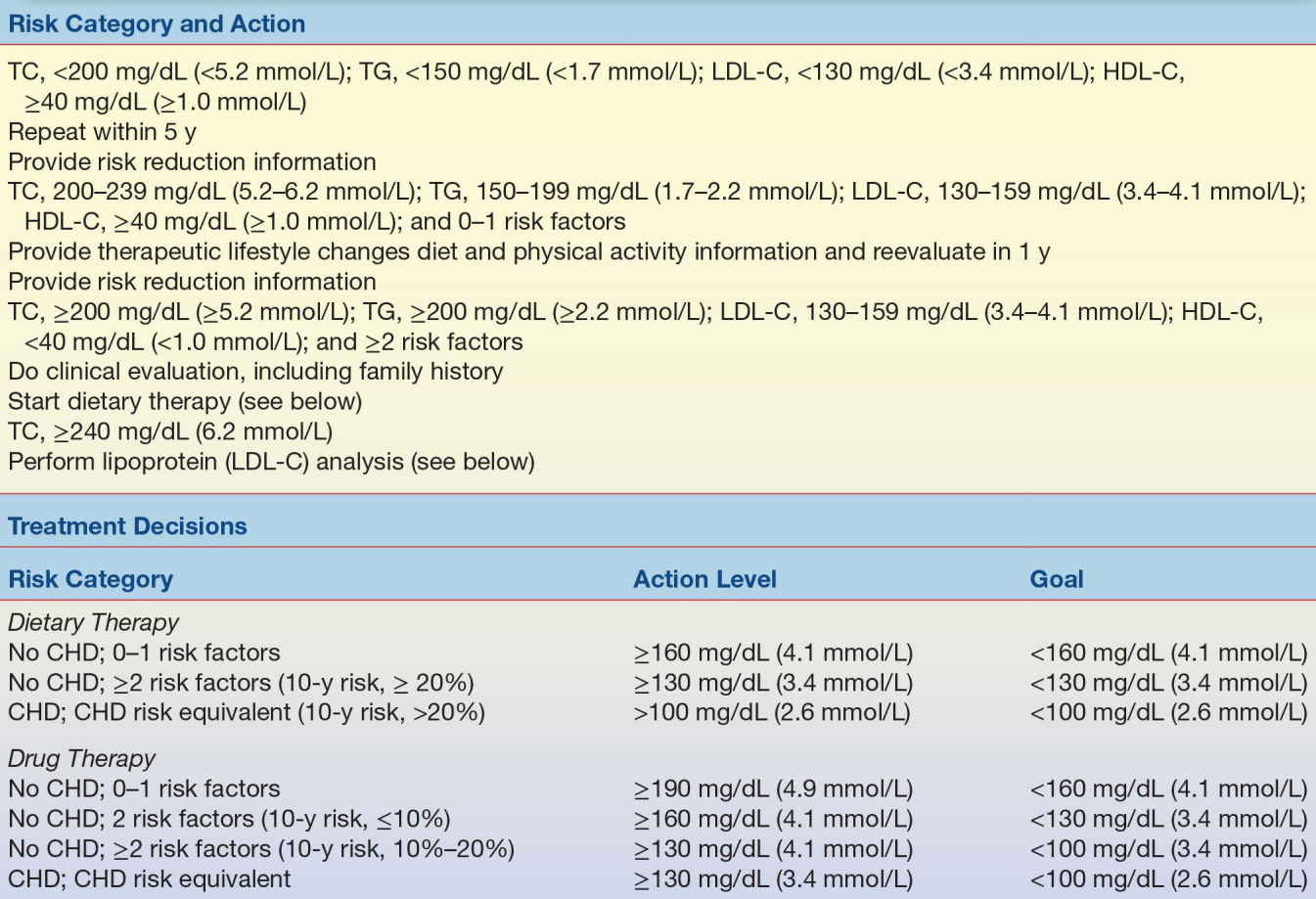
CHD, coronary heart disease; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; TC, total cholesterol; TG, triglycerides.
Classic bile acid sequestrant drug treatments, such as cholestyramine, colesevelam, and colestipol, work by sequestering cholesterol in the gut so that it is not absorbed; this action enhances conversion of cholesterol into bile acids in the liver, reduces hepatic cholesterol content, and enhances the activity of LDL receptors, and until recently, these were considered to be the only safe drugs for use in children.48 These bile acid sequestrants lower LDL-C concentrations by 15% to 30% but may increase triglycerides.52 It is important to note that these agents interfere with the absorption of fat-soluble vitamins and some drugs like digoxin, warfarin, and thyroid supplements, resulting in altered blood concentrations of these drugs and prothrombin times. Bile acid sequestrants are contraindicated in individuals with triglyceride levels greater than 400 mg/dL (4.5 mmol/L). Bile acid sequestrants have uncomfortable adverse effects such as bloating and constipation and are poorly tolerated.
The most effective class of drugs for managing patients with dyslipidemia are the HMG-CoA reductase drug inhibitors, such as lovastatin, atorvastatin, and rosuvastatin. These drugs, commonly known as statins, block intracellular cholesterol synthesis by inhibiting HMG-CoA reductase, a rate-limiting enzyme in cholesterol biosynthesis. The reduced level of cholesterol in hepatocytes increases the expression of the LDL receptor, which removes LDL from the circulation, thus reducing the deposition of LDL into vessels and the formation of plaques. All statins are probably identical in their main mechanism but differ in dose–response curves. The major safety issues with statins are myositis and hepatotoxic effects; however, patient monitoring in clinical trials has shown that fewer than 2% of patients have sustained increases in liver enzymes. Routine monitoring of serum transaminases is recommended and creatine kinase for patients complaining of muscle-related symptoms. These drugs typically reduce LDL-C by as much as 20% to 40%, raise HDL-C by 5% to 10%, and can lower triglyceride by 7% to 43%, depending on the initial triglyceride level.45,53 Statins are generally well tolerated, with maximum effects observed after 4 to 6 weeks.45,53 Clinical trials have shown that statin is beneficial for both the primary and secondary prevention of CHD.31
Niacin or nicotinic acid at high doses (~2 to 6 g/d) is also a potent drug for reducing LDL-C levels (by 5% to 25%) and is the only effective drug at this time for significantly raising HDL-C levels (by 15% to 35%).54 Niacin can also lower triglyceride levels by 20% to 50%55; however, it causes flushing, pruritus, hyperuricemia, and diarrhea in many patients and can be hepatotoxic.56 The precise mechanism of action for niacin is not known, although it appears to act in the liver to reduce the formation of LDL and VLDL by inhibiting lipolysis of triglyceride in adipose tissue and triglyceride synthesis in the liver. This action is sufficient to control severe hypertriglyceridemia. Newer slow-release formulations of niacin and combination drugs reduce incidence of flushing.57
Fibric acid derivatives, such as clofibrate, gemfibrozil, fenofibrate, and etofibrate, are most often used to reduce triglyceride by 20% to 50% and increase HDL-C levels by 10% to 20%.50,56 The effects of fibric acid derivatives on LDL-C concentrations are less predictable and depend on the individual’s level of triglyceride. Individuals receiving fibric acid agents should have liver function tests monitored, because these drugs are associated with elevations in transaminases, as well as creatine kinase for patients complaining of muscle ache. Patients on fibrates and warfarin should also have their prothrombin time monitored, because fibrates can displace warfarin from their protein-binding sites.52
Ezetimibe is a new drug that inhibits the absorption of cholesterol by inhibiting the Niemann-Pick C1-like 1 (NPC1-L1) transporter in the intestine without impacting the absorption of fat-soluble nutrients. Ezetimibe therapy has been shown to decrease LDL-C concentrations by approximately 20%.58 Ezetimibe in conjunction with statins results in greater reductions in LDL-C concentration than with statin alone. As with the use of statin drugs, liver function tests should be performed on individuals taking ezetimibe.58
Fish oil products that contain omega-3 fatty acids, eicosapentaenoic and docosahexaenoic acids, can be used to lower triglyceride levels (up to 45%) and raise HDL-C (up to 13%) in patients, although the mechanism of action is not fully understood; it has been shown that fish oils decrease hepatic triglyceride synthesis59 via inhibition of diacylglycerol acyltransferase, fatty acid synthase, and acetyl-CoA carboxylase enzyme activities. Fish oils also enhance fatty acid β-oxidation by stimulating peroxisome proliferator–activated receptors. In addition, fish oils decrease the hepatic pool of triglycerides by suppressing transcription of the SREBP-1c gene, thereby inhibiting de novo synthesis of fatty acids and triglycerides.60 However, elevations in LDL-C concentrations (up to 40%) have been observed in some patients receiving omega-3 fatty acids,61 but LDL-C changes from small, dense atherogenic particles to larger, more buoyant and less atherogenic particles. Also, the rise in LDL-C is usually less than the decrease in VLDL cholesterol (VLDL-C). In addition, elevations in serum transaminases have been reported and these enzymes should be monitored periodically for those patients taking omega-3 fatty acids.
Hyperlipoproteinemia
Disease states associated with abnormal serum lipids are generally caused by malfunctions in the synthesis, transport, or catabolism of lipoproteins. Dyslipidemias can be subdivided into two major categories: hyperlipoproteinemias, which are diseases associated with elevated lipoprotein levels, and hypolipoproteinemias, which are associated with decreased lipoprotein levels. The hyperlipoproteinemias can be subdivided into hypercholesterolemia, hypertriglyceridemia, and combined hyperlipidemia, with elevations of both cholesterol and triglycerides.
Hypercholesterolemia
Hypercholesterolemia is the lipid abnormality most closely linked to heart disease.62 One form of the disease, which is associated with genetic abnormalities that predispose affected individuals to elevated cholesterol levels, is called FH. Homozygotes for FH are fortunately rare (1:1 million in the population) and can have total cholesterol concentrations as high as 800 to 1,000 mg/dL (20 to 26 mmol/L). These patients can have their first heart attack when still in their teenage years.63 Heterozygotes for the disease are seen much more frequently (1:500 in the population), because it is an autosomal codominant disorder; a defect in just one of the two copies of the LDL receptor can adversely affect lipid levels. Heterozygotes tend to have total cholesterol concentrations in the range of 300 to 600 mg/dL (8 to 15 mmol/L) and, if not treated, become symptomatic for heart disease in their twenties to fifties. Approximately 5% of patients younger than age 50 with CAD are FH heterozygotes. Other symptoms associated with FH include tendinous and tuberous xanthomas, which are cholesterol deposits under the skin, and arcus, which are cholesterol deposits in the cornea.64
In both homozygotes and heterozygotes, the cholesterol elevation is primarily associated with an increase in LDL-C. These individuals synthesize intracellular cholesterol normally but lack, or are deficient in, active LDL receptors. There are several classes of defects in the LDL receptor gene that are associated with FH. Class 1 mutations result in null allele where no LDL receptor protein is identified. Class 2 mutation, which accounts for the majority of clinical mutations in FH, creates a membrane protein that is synthesized but not transported to the Golgi efficiently and, thus, is degraded. Class 3 mutations result in an LDL receptor that cannot bind LDL. Class 4 mutations create an LDL receptor that cannot internalize the LDL and transport it to the Golgi, and class 5 mutations create LDL receptors that do not recyle.65 Mutations in the LDL receptor gene promoter region have also been described. Consequently, LDL builds up in the circulation, because there are insufficient receptors to bind the LDL and transfer the cholesterol into the cells. Cells, however, which require cholesterol for use in cell membrane and hormone production, synthesize cholesterol intracellularly at an increased rate to compensate for the lack of cholesterol from the receptor-mediated mechanism.
CASE STUDY 15.3
A 43-year-old white man was diagnosed with hyperlipidemia at age 13 years, when his father died of a myocardial infarction at age 34 years. The man’s grandfather had died at age 43 years, also of a myocardial infarction. Currently, the man is active and asymptomatic with regard to CHD. He is taking 40 mg of lovastatin (Mevacor), two times per day (maximum dose). He had previously taken niacin but could not tolerate it because of flushing and gastrointestinal distress, nor could he tolerate cholestyramine resin (Questran). His physical examination is remarkable for bilateral Achilles tendon thickening/xanthomas and a right carotid bruit (see Case Study Table 15.3.1).
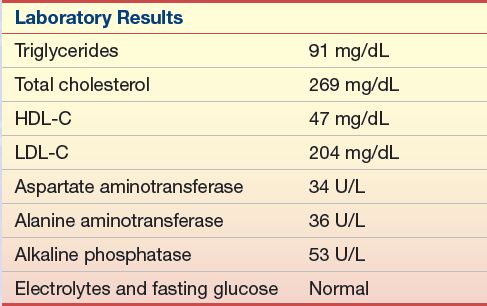
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


