Cardiovascular disease (CVD) commonly occurs in the general population and affects the majority of people older than 60 years. This includes four major types of CVD based on the location in which it occurs: (1) coronary heart disease (CHD), (2) cerebrovascular disease, (3) peripheral arterial disease, and (4) aortic atherosclerotic disease. CHD manifests as myocardial infarction (MI) (heart attack), angina pectoris (chest pain), heart failure, and sudden cardiac death. Cerebrovascular disease manifests as a stroke or transient ischemic attack (short, “reversible strokes”), while peripheral arterial disease manifests by intermittent claudication (acute localized pain to the arms and legs). Aortic atherosclerosis manifests as aneurysms (abnormal widening of an artery) or dissection (tears in thoracic or abdominal aorta), which are acute life-threatening events. There are common themes in the pathogenesis of these CVDs, including the presence of atherosclerosis, which increases the likelihood of ischemia (lack of blood supply) localized to different parts of the body. CHD, in which atherosclerosis and ischemia are localized to the vasculature of the heart, accounts for one-third of the total cases of CVD. The critical role of laboratory testing in diagnosing CHD will be the focus of this chapter.
One of the most common reasons people go to the emergency department is for the acute onset of chest pain. While nearly seven million people end up in emergency departments for this reason, only 15% to 25% of them actually have acute coronary syndrome (ACS) resulting from CHD.1,2 There are a number of noncardiac reasons for having chest pain, including common maladies such as esophageal reflux (“heartburn”) due to stomach acid moving retrograde into the esophagus. It is critical to differentiate cardiac from noncardiac causes, but differentiating patients with ACS or other life-threatening conditions from patients with non–life-threatening diseases from noncardiac causes is challenging. This is evidenced by the fact that the diagnosis of ACS is missed in approximately 2% of patients, which results in substantial morbidity and mortality.2 Several advances in risk-stratifying patients with chest pain have been made, including better laboratory markers of cardiac injury,3 which allows for rapid identification of those patients whose chest pain is due to CHD. Such patients benefit from rapid initiation of therapy directed at ACS. Patients without elevated cardiac markers are typically at lower risk and may undergo further evaluation of the cause of their symptoms, including through radionuclide imaging and computed tomography (CT).
The lack of an adequate blood supply to the heart leads to ischemia. Since the heart depends upon a constant supply of oxygen and nutrients supplied by the coronary arteries, any disruption in the balance of oxygen supply and cardiac demand puts the heart at serious risk. Adequate supply of blood to the heart is compromised by a number of factors, most commonly the formation of a thrombus in the context of underlying atherosclerosis, which is the underlying cause for MI (narrowing of the coronary arteries). Atherosclerosis is a chronic process involving damage to the endothelium and the buildup of cholesterol-rich lesions that threaten to occlude the vasculature.
CARDIAC ISCHEMIA, ANGINA, AND HEART ATTACKS
Cardiac ischemia can lead to activity-related chest pain (stable angina) or an ACS. Stable angina predictably occurs with a given amount of activity and resolves with rest, whereas ACS frequently occurs unpredictably and does not respond to cessation of activity. ACS is further classified as unstable angina or the more severe acute MI, also known as a heart attack. Cardiac biomarkers are normal in the setting of unstable angina, but elevated in MI. The “classic” manifestation of cardiac ischemia is angina, described as a squeezing of the chest, heavy chest pressure, a burning feeling, or difficulty breathing.4 This “classic” manifestation often radiates to the left shoulder, neck, or arm and typically increases in intensity over a period of minutes and gets worse with either physical or psychological stress. However, the symptoms often arise in the absence of situations that precipitate it. Nonclassic symptoms, more commonly experienced by women, include a more stabbing, pulsating, or sharp chest pain rather than the typical viselike pressure, nausea, shortness of breath, or abdominal pain.5, 6, 7 In addition to these heart-related symptoms, the American Heart Association (AHA) and the American College of Cardiology (ACC) have also defined chest-related symptoms that are not indicative of cardiac involvement (see Table 26.1).

Guidelines from the ACC and the AHA categorize patients into two classes: class I, patients with suspected ACS and high-risk features (chest pain, syncope/presyncope, or palpitations), and class IIb, patients with less severe symptoms.8 Class I patients should be immediately transported to ED by emergency medical services.8 Class IIb patients may be considered for referral to the ED, a chest pain unit, or a facility capable of performing evaluation.8 If a patient does not need immediate intervention because of circulatory collapse (i.e., the patient is conscious), a history is taken to evaluate the chest pain for its likelihood of being an MI. Risk factors for atherosclerosis should also be solicited, including advanced age, male gender, diabetes, and any previous history of MI. Younger patients with a low risk of ACS should be carefully screened for cocaine use, as it can cause vascular spasm of the coronary arteries, resulting in ischemia and chest pain.8 Patients with chest pain are initially evaluated by a physical examination, an electrocardiogram (ECG), a chest x-ray, and the detection of biomarkers by laboratory tests. The physical examination identifies possible causes of precipitating myocardial ischemia (high blood pressure, pulmonary disease, and heart failure or cardiac valve disease)8 and may identify a noncardiac cause for the symptom. An ECG should be obtained within the first 10 minutes after presentation for further risk stratification. The presence of new or transient changes (>1 mm) in the ST segment of the ECG is strongly suggestive of acute ischemia.8 Less specific ST-segment changes or T-wave abnormalities can also be helpful in risk-stratifying patients.8 A normal ECG, however, does not rule out the presence of ACS. Chest radiography can also assist in the evaluation of a patient with chest pain by identifying a noncardiac source of the pain (e.g., pneumonia, pneumothorax, and aortic dissection) or sequelae of underlying cardiac causes (e.g., pulmonary edema caused by cardiac dysfunction).8
The use of biomarkers is critical in the evaluation of acute chest pain to identify angina (chest pain) as an MI (heart attack). Universal definitions of MI were recently published in 2008 outlining the consensus reached by the joint European Society of Cardiology/ACC/AHA/World Health Federation Task Force (see Table 26.2). These guidelines emphasized the use of the term MI only in the setting of myocardial ischemia and not as a result of any other cause9 and designated several types of MI (Table 26.3). The 2014 ACC/AHA guidelines emphasize cardiac troponins as the most sensitive and specific biomarkers for non–ST-segment elevation ACS.8
TABLE 26.2 Universal Definition of MI Released by the Joint European Society of Cardiology/American College of Cardiology/American Heart Association/World Health Federation Task Force (2007)9

ECG, electrocardiogram; PCI, percutaneous coronary intervention (also known as “coronary angioplasty” or “angioplasty”); CABG, coronary artery bypass graft (also known as “bypass surgery”); URL, upper reference limit.
TABLE 26.3 Classification of MI According to the Universal Definition of Myocardial Infarction Released by the Joint European Society of Cardiology/American College of Cardiology/American Heart Association/World Health Federation Task Force (2007)9

MI, myocardial infarction; LBBB, left bundle branch block; PCI, percutaneous coronary intervention (also known as “coronary angioplasty” or “angioplasty”); CABG, coronary artery bypass graft (also known as “bypass surgery”).
CASE STUDY 26.1
A 15-month-old girl with a heart murmur since birth was evaluated for repeated pulmonary infections, failure to grow, cyanosis, and mild clubbing of fingers and toes. She had been on digitalis therapy by the referring physician. The radiograph showed a moderately enlarged heart and an enlarged pulmonary artery. Pertinent laboratory data were obtained.

A cardiac catheterization was performed, and a large ventricular septal defect was found.
questions
1. How does this congenital defect affect the body’s circulation?
2. Why are the red cell measurements increased in this patient?
3. What treatment will be suggested for this patient?
4. What is this patient’s prognosis?
THE PATHOPHYSIOLOGY OF ATHEROSCLEROSIS, THE DISEASE PROCESS UNDERLYING MI
Atherosclerosis is a chronic disease process that occurs over a number of years and contributes to approximately 50% of all deaths in modern Western societies.10 Evidence of atherosclerosis can often be found in the human aorta before the age of 10,11 but atherosclerosis becomes pathologic with the development of atherosclerotic plaques (atheromas), which predispose the vasculature to thrombosis, leading to organ ischemia and infarction (Fig. 26.1). The pathophysiology of atherosclerosis is gradual and complicated, involving a progressive accumulation of lipids, smooth muscle cells, macrophages, and connective tissue within the intima of large- and medium-sized arteries, ultimately causing luminal narrowing and decreased perfusion (Fig. 26.1). Although the exact etiology of atherosclerosis remains unclear, the reaction to injury hypothesis is strongly favored by current evidence, proposing that atherosclerosis is due to a chronic inflammatory response to an accumulation of subtle vascular wall injuries.12
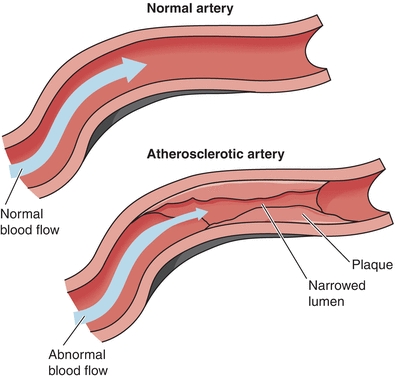
FIGURE 26.1 Comparison of normal and atherosclerotic arteries. Narrowing the arterial lumen due to atherosclerotic plaque leads to abnormal blood flow, contributing to progression of atherosclerosis. (Adapted from NHLBI Atherosclerosis homepage, http://www.nhlbi.nih.gov/health/health-topics/topics/atherosclerosis/)
Pathohistological evidence from human and experimental studies demonstrates that endothelial and inflammatory cells interact with chemical and inflammatory mediators to promote the development of atherosclerotic plaques. This process begins with vascular injury, which is initiated when endothelial cells are damaged or rendered dysfunctional by vascular abnormalities, such as turbulent blood flow, hyperlipidemia, and hyperhomocysteinemia. A damaged vascular endothelium has an increased permeability to circulating lipids, so having high cholesterol (hyperlipidemia) in the wake of a damaged endothelium favors the accumulation of lipoproteins, predominantly low-density lipoprotein (LDL), and very–low density lipoprotein (VLDL), within the arterial intima (Fig. 26.1).13 In addition, apoB-containing LDL has high affinity for arterial wall proteoglycans.14 Retention of lipids in arterial walls due to hypercholesterolemia and/or elevated levels of apoB-containing lipoproteins is thus a crucial step in the pathogenesis of atherosclerotic lesions. The central role of cholesterol accumulation (i.e., LDL and VLDL) in the progression of atherosclerosis is the reason treating high cholesterol is such a priority in preventing and attenuating heart disease.
Once lesion initiation has begun, LDL deposited within the intima is oxidized by endothelial cells, lipoxygenase, and free radicals generated by the auto-oxidation of homocysteine.11 The formation of oxidized LDL is central to lesion progression. Oxidized LDL is toxic to endothelial cells, causing additional intimal damage and subsequent retention of cholesterol-rich lipoproteins, and it elicits an inflammatory response that releases proinflammatory cytokines and recruits inflammatory cells to the early lesion. Among the earliest recruited leukocytes are neutrophils and monocytes, which play critical roles in early atherogenesis by maintaining a proinflammatory state around the initial lesion (Fig. 26.2).10
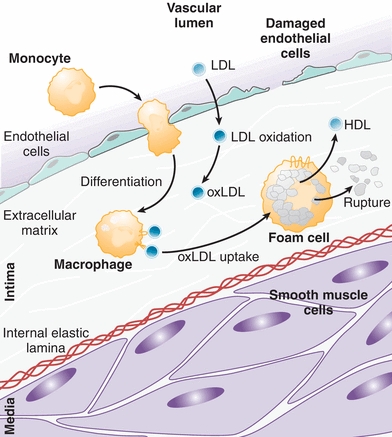
FIGURE 26.2 LDL deposition and oxidation within the vessel wall leads to monocyte recruitment and differentiation into activated macrophages that phagocytose oxidized LDL (oxLDL) to become foam cells. Foam cells release HDL and proinflammatory mediators, and their rupture contributes to lesion progression. (Adapted from Glass CK, Witztum JL. Atherosclerosis: the road ahead. Cell. 2001;104(4):503–516, Ref.10.)
Maturation of monocytes into activated macrophages is a key step in lesion progression. Macrophage scavenger receptors recognize oxidized LDL, but not native LDL, and activated macrophages rapidly phagocytose cholesterol-rich lipoproteins that have been oxidized within the vessel wall (Fig. 26.2).11 Excessive uptake of oxidized LDL transforms macrophages into bloated, cholesterol-filled cells called foam cells.15 Filled with cytoplasmic lipid droplets, foam cells exhibit a variety of functions that both promote lesion progression, including the production of proinflammatory signals, and counter lesion progression, such as the secretion of HDL (Fig. 26.2). Foam cells also display numerous metabolic abnormalities, including activation of inflammasomes and the NF-kB pathway, and it is generally agreed that foam cells play a harmful role in lesion progression.15 In addition, rupture of foam cells and release of their contents cause further damage to the vascular endothelium, stimulating more inflammation. Intimal deposition and subsequent oxidation of LDL and its effects on monocyte differentiation are thus of key importance in the early development of an atherosclerotic lesion.
As the cycle of endothelial cell damage progresses, additional cell types are recruited to the plaque, in particular T and B lymphocytes and macrophages (Fig. 26.3). These cells are activated by a stream of cytokines, such as interleukin (IL)-1 and tumor necrosis factor-α, that are released by endothelial cells within the plaque.16 Interactions between T cells and foam cells promote a chronic inflammatory state and help recruit smooth muscle cells into the intima (Fig. 26.3).10 Additionally, growth factors, such as platelet-derived growth factor, fibroblast growth factor, and tissue growth factor-α, are released by lymphocytes and endothelial cells, further stimulating smooth muscle cell migration and activation.16
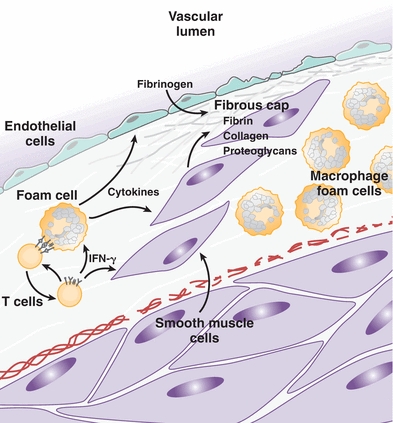
FIGURE 26.3 T cells and foam cells maintain a proinflammatory state that induces the migration of smooth muscle cells into the intima, where they secrete collagen, proteoglycans, and fibrin that form a fibrous cap around the atheroma. (Adapted from Glass CK, Witztum JL. Atherosclerosis: the road ahead. Cell. 2001;104(4):503–516, Ref.10.)
Once smooth muscle cells migrate into the core of the atheroma, they proliferate and deposit extracellular matrix components that give stability and strength to the plaque.10 Cytokines released from T cells, such as interferon-γ, exert a number of both pro- and antiatherogenic effects on macrophages and smooth muscle cells, but the net effect of this cytokine release is proinflammatory and proatherogenic. Smooth muscle cells are induced to secrete collagen, elastin, and proteoglycans that form a fibrous backbone and outer shell that fix the plaque firmly in the vessel wall (Fig. 26.3). As the atheroma grows, its core becomes increasingly isolated from surrounding blood supply, leading to hypoxia that stimulates release of proangiogenic cytokines. This causes aberrant neovascularization around the periphery of the plaque, which predisposes to hemorrhage and deposition of erythrocyte components, such as hemosiderin and lipids, within the plaque core.17 Microvasculature hemorrhage and subsequent deposition of additional debris provokes further inflammation, leukocyte recruitment, and remodeling of the plaque.
The steps described above—beginning with lipid and cellular infiltration following vascular injury and progressing to chronic inflammation and fibrosis—continue in a feed-forward cycle that ultimately culminates in complete vessel occlusion, thrombosis, plaque rupture, or some combination of the three. Each of these possible outcomes predisposes to organ ischemia. In the context of the heart, coronary artery ischemia and resultant hypoxia pose an immediate risk of cellular injury due to the high metabolic activity and oxygen demand of the myocardium. The ultimate outcome of such injury is ischemic heart disease, varying in severity from angina to MI, the symptoms of which are dependent on the extent of coronary artery atherosclerosis. The most common locations for symptomatic cardiac atheroma formation are the proximal left anterior descending, the proximal left main coronary, and the entire right coronary arteries. Coronary atherosclerosis typically becomes symptomatic only after atherosclerotic plaques obstruct approximately 75% (or >50% in the left main) of the cross-sectional area of the vessel.10
MARKERS OF CARDIAC DAMAGE
Initial Markers of Cardiac Damage
The first biochemical markers of cardiac damage were discovered in 1954 when Karmen et al.18 hypothesized that “destruction of cardiac muscle, reported rich in transaminase activity, might result in a release of this enzyme into the blood stream and might thus increase the serum transaminase activity.” Today, serum biomarkers have become the centerpiece of evaluation and management of patients presenting with chest pain. The underlying principle behind serum biomarkers of cardiac damage relies on the fact that cell death releases intracellular proteins from the myocardium into the circulation. Detection of cardiac proteins in plasma provides insight into the occurrence, extent, and timing of MI, all of which are critical for proper medical management.
The first cardiac markers to be used extensively in clinical practice were glutamic oxaloacetic transaminase, lactic dehydrogenase, and malic dehydrogenase. The first, glutamic oxaloacetic transaminase, now known as aspartate transaminase (AST), became widely used in the diagnosis of MI shortly after its discovery as a serum marker by Ladue et al. in 1954.19 But because of the high false-negative rate of AST, the labor-intensive nature of the AST assay, and the short window of AST elevation, AST was soon replaced by lactate dehydrogenase (LD) as the marker of choice.20
Compared with AST, LD was found to be a more sensitive marker of MI that remains elevated for significantly longer post-MI, up to 2 weeks. But LD is involved in NADH-dependent reactions in the glycolytic pathway, which takes place in nearly all cells in the body, and its specificity to cardiac muscle is thus quite low. Early researchers observed that serum LD levels were elevated in other conditions such as cancer and anemia.20 These pitfalls were overcome to some extent through assays that differentiate between LD of cardiac and noncardiac origin.21 Specifically, five different isoforms of LD (LD1 to LD5) are found in human plasma,22 which correspond to the organ from where the enzyme originates. LD1 is most abundant in the myocardium, and LD5 is expressed mainly in the skeletal muscle and liver. Therefore, the diagnosis of MI was made by comparing plasma LD1 and LD2 levels. Because LD1 is specific to the myocardium, plasma normally contains greater levels of LD2 than LD1; however, damage to the myocardium and subsequent release of LD1 can cause plasma LD1 levels to surpass LD2.23 Early studies demonstrated that the plasma LD1:LD2 ratio exceeds approximately 0.75 24 to 48 hours past the onset of symptoms of MI and remains elevated for up to 2 weeks.23 The LD1:LD2 ratio was thus of great utility as an early biomarker in the management of patients presenting several days after possible MI.
Creatine kinase (CK) was the next marker to gain favor. Like LD, CK is found in nearly all cells in the body, but unlike LD, CK catalyzes a reaction important for high-energy phosphate production (the conversion of creatine to creatine phosphate), which is greatly upregulated in the muscle cells and brain. High levels of CK are thus found in all muscle cells, in particular striated muscle. Damage to the muscle or brain results in rapidly detectable increases in plasma CK, which can be readily detected in plasma with a high sensitivity at short times after muscle injury. After the discovery in 1960 of elevated levels of plasma CK post-MI,24 CK became an important marker of cardiac damage. In a typical patient with acute MI, serum CK levels were found to exceed the normal range within 6 to 8 hours, to reach a peak of two- to tenfold normal by 24 hours, and then decline to the normal range after 3 to 4 days (Fig. 26.4).26 A CK plasma concentration greater than two times normal was shown to correlate with MI,25,27 and an elevation in serum CK, together with elevated AST and LD, was used as the primary means of enzymatic detection of MI for many years.
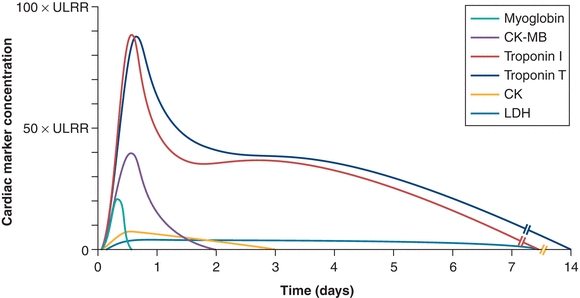
FIGURE 26.4 Temporal elevations of important markers of myocardial damage post-MI. ULRR, upper limit of reference range. (Adapted from French JK, White HD. Clinical implications of the new definition of myocardial infarction. Heart. 2004;90(1):99–106, Ref.25.)
Unfortunately, the ubiquitous expression of CK in all striated muscle was a problem for specificity of CK as a marker for MI, in spite of its high sensitivity. CK elevations were readily detected in conditions such as stroke, pulmonary disease, and chronic alcoholism and after strenuous exercise.28 This obstacle was initially overcome through the discovery that CK exists in three cytoplasmic isoenzymes. The cytoplasmic isoenzymes are dimers composed of combinations of M and B subunits (where M is for muscle and B is for brain). Dimers of 2 M subunits are called CK-MM, 2 B subunits as CK-BB, and 1 M and 1 B subunit as creatine kinase MB (CK-MB). Elevated plasma levels of CK-MM or CK-BB can be found after injury to the muscle or brain, respectively. After it was found that 15% to 30% of CK in the myocardium is MB, compared with 1% to 3% in normal striated muscle, detection of elevated CK-MB was shown to be highly specific to myocardial damage.29 Additionally, elevations in serum CK-MB could be detected at 4 to 6 hours after the onset of MI symptoms, significantly less than the 24 to 48 hours necessary to detect peak LD plasma levels (Fig. 26.4). Like its rapid rise post-MI, CK-MB levels quickly drop to baseline levels by 2 to 4 days post-MI, compared with 10 to 14 days for LD.30 Because of its higher specificity and its rapid elevation post-MI, CK-MB was long considered the most reliable serum marker of MI and is still widely used today.31,32
Use of these three markers—AST, LD, and CK—was the cornerstone of post-MI management for several decades. Each had its individual applications: CK for the early presentation, LD for the late presentation, and AST for the intermediate.33 But their lack of specificity to the myocardium and to myocardial injury fueled the search for better markers. Extensive research in the 1970s led to the discovery that muscle cells express structural and regulatory proteins, such as troponin and tropomyosin, in a pattern that is tissue specific.32 The subsequent development of rapid and sensitive techniques for detection of the tissue-specific forms of troponin quickly made troponin a promising marker of cardiac injury.34
Cardiac Troponins
Troponin is a complex of three proteins that regulate the calcium-dependent interactions of myosin heads with actin filaments during striated muscle contraction. Troponin T (TnT) binds the troponin complex to tropomyosin, troponin I (TnI) inhibits the binding of actin and myosin, and troponin C (TnC) binds to calcium to reverse the inhibitory activity of TnI.35 The troponin complex is responsible for transmitting the calcium signal that triggers muscle contraction (Fig. 26.5). Several properties of troponin made it attractive as a marker of myocardial damage.
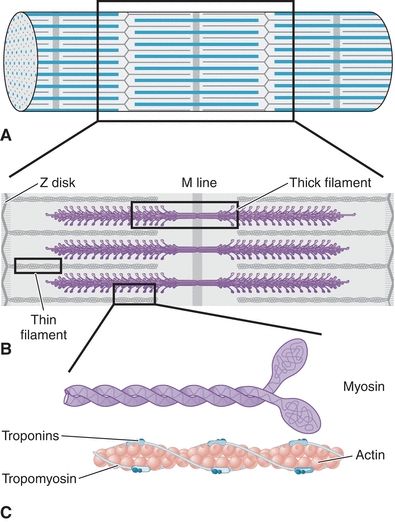
FIGURE 26.5 Regulatory proteins in the contractile apparatus of striated muscle. Striated muscle is composed of bundles of myofibril-containing fibers, made up of repeating units of cross-striations called sarcomeres (A). The sarcomere is composed of thick and thin myofilaments, which tether the Z disk to the M line (B). Thick filaments are composed primarily of large molecules of myosin, which have a rodlike tail region and a protruding head region (C). Thin filaments are composed of actin, which plays a structural role, and troponin and tropomyosin, which line actin filaments and regulate calcium-dependent interactions with myosin on thick filaments to stimulate contraction.
In contrast to other cardiac markers, troponins were found to have tissue-specific isoforms that could be used to detect damage to the heart. Although the same isoform of TnC is expressed in slow-twitch (type 2) and cardiac muscle, unique isoforms of TnI and TnT are expressed in fast-twitch (type 1), slow-twitch, and cardiac muscle.35 Each TnI, TnT, and TnC isoform is encoded by a separate gene and expressed in a muscle-type–specific manner.36 This allows for highly specific detection of troponin of cardiac origin (e.g., cardiac TnI [cTnI]). Further increasing the sensitivity of troponin detection was the demonstration that cardiac troponins are very tightly complexed to the contractile apparatus, such that circulating levels of cardiac troponins are normally extremely low.35 Early studies demonstrated that normal levels of circulating cTnI are below 10 ng/mL, but patients with acute MI can have serum cTnI levels well over 100 ng/mL,37 demonstrating the specificity of cardiac troponins as indicators of myocardial cell damage.
The development of sensitive detection methods was the next step toward integrating cardiac troponins into the diagnostic workup of MI. Because the skeletal and cardiac isoforms of TnT and TnI contain differing amino acid sequences, monoclonal antibodies can be raised against cardiac-specific epitopes. This allowed for the development of sensitive immunoassays for cardiac troponins that have a far greater specificity than other markers. For example, detection of cTnI was specific for cardiac injury even in acute muscle disease, chronic muscle disease, chronic renal failure, and following intense exercise, such as running a marathon.38 Additionally, measurement of plasma troponin levels allows for the detection of myocardial cell injury in syndromes, such as acute ischemic syndrome or myocarditis, that is undetectable using other markers.39
Finally, troponins offer additional advantages due to the timing of their release. CK-MB levels elevate rapidly post-MI (4 to 6 hours) but return to baseline after 2 to 4 days; CK-MB can thus only be used within a short window of time after a suspected MI. Conversely, LD levels remain elevated for up to 1 week but are not detectable until 24 to 48 hours post-MI (Fig. 26.4). Cardiac troponins, on the other hand, are detectable in the plasma at 3 to 12 hours after myocardial injury, peaking at 12 to 24 hours and remaining elevated for more than 1 week: 8 to 21 days for TnT and 7 to 14 days for TnI (Fig. 26.4).39 Cardiac troponins thus offer the widest window for detection post-MI and with the highest sensitivity and specificity.
Taken together, the characteristics of specific, sensitive, and rapid detection propelled cardiac troponins to the center of MI diagnosis, where they still remain. Cardiac troponin (I or T) is currently the preferred biomarker for myocardial necrosis, recognized to have nearly absolute myocardial tissue specificity as well as high clinical sensitivity, detecting even minor cardiac damage (as utilized in the universal definition of MI).9 An elevated plasma cardiac troponin level is defined as a measurement greater than the 99th percentile of a normal reference value range, specified as the upper limit of the reference range (Fig. 26.4). Current guidelines recommend drawing blood for the measurement of troponin as soon as possible after onset of symptoms of a possible MI (universal definition of MI).9 If troponin assays are not available, the next best alternative is CK-MB, and measurement of total CK is no longer recommended for the diagnosis of MI for the reasons outlined above (universal definition of MI).9
CK-MB and Troponin I/Troponin T Considerations in Kidney Disease Patients
Prognosis in chronic kidney disease. Increases in cTnI/cTnT are associated with increases in short-term cardiac outcomes in patients with chronic kidney disease diagnosed with acute MI.40, 41, 42 Stably increased serum troponin levels predict worse long-term cardiovascular outcomes and worse survival in asymptomatic CKD patients in the absence of acute MI.43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54
Dialysis patients. In a recent meta-analysis of 11 studies, elevated cTnT was associated with an increase in all-cause mortality (95% confidence interval [CI] 2.4 to 43).42 Few studies here used the high-sensitivity assay; cTnT measured with high-sensitive assay has also been reported as an independent predictor of all-cause mortality in dialysis patients.55,56
Effect of hemodialysis. In kidney failure, the need to dialyze patients is common when failure is severe. Hemodialysis involves filtering the blood to remove the built-up toxins and results in altering the concentration of cardiac enzymes. Studies have yielded conflicting results. Dialysis minimally changes CK-MB and did not change cTnI concentrations.57 Decreased cTnI and cTnT levels decreased 27% to 37% following dialysis with a high-flux membrane (but not low-flux membrane).58 cTnT rose after dialysis in patients with CVD, suggesting the rise in cTnT resulting from cardiac injury.59 While the cut-offs for the diagnosis for AMI should be the same for dialysis patients and nondialysis chronic kidney disease patients, considerations above are important with the growing number of patients surviving with chronic kidney disease.
Other Markers of Cardiac Damage
In spite of the nearly universal use of troponin and CK-MB in diagnosing cardiac damage, it is worthwhile mentioning additional markers of cardiac damage that have been used in the past and/or proposed experimentally. These include myoglobin, heart-type fatty acid–binding protein (H-FABP), ischemia-modified albumin (IMA), myeloperoxidase (MPO), C-reactive protein (CRP), myeloid-related protein (MRP)-8/14, and pregnancy-associated plasma protein A (PAPP-A). While their clinical utility has not been universally established, they are discussed frequently as potential alternatives, so they have been discussed here briefly.
CASE STUDY 26.2
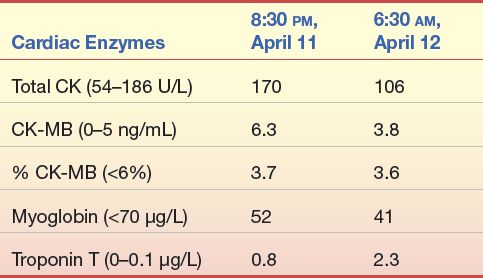
A 59-year-old woman came to the emergency department of a local hospital complaining of pain and a feeling of heaviness in her abdomen for several days. She reported no weakness, chest pain, or left arm pain. She has no chronic health problems except for seasonal allergies and slightly elevated total cholesterol.
Questions
1. Do the symptoms and personal history of this patient suggest acute MI?
2. Based on the preceding laboratory data, would this diagnosis be acute MI?
3. Why or why not?
Myoglobin
Myoglobin is an iron- and oxygen-binding protein found exclusively in the muscle and is normally absent from the circulation. The myoglobin found in the muscle forms pigments, giving the muscle its characteristic red color. The exact color of the muscle depends on the oxidation state of the iron atom in myoglobin and the amount of oxygen attached to it. Myoglobin is a small protein, which is released quickly when the muscle is damaged (see Fig. 26.4 for serum release pattern).60 It has a very short half-life of 9 minutes.61 Because myoglobin is released so quickly, it has been proposed as an adjunct marker for troponin or CK-MB in the early diagnosis of MI.62 There are limitations to using myoglobin as a marker of cardiac damage. It is not specific for the heart, being elevated with any cause of skeletal muscle damage, and is cleared in an irregular pattern.63 Recent studies have also shown that high-sensitivity troponin assays can detect elevated troponin levels prior to elevations in myoglobin,64, 65, 66 making myoglobin’s use less attractive.
Heart-Type Fatty Acid–Binding Protein
H-FABP is a small protein, which behaves much like myoglobin in its kinetics and release. It is found in all muscles, but in contrast to myoglobin, H-FABP is found relatively more abundantly in the heart.67 The utility of H-FABP has been described for the diagnosis of early acute MI. The sensitivity for the diagnosis of an acute MI has been reported to be higher than cardiac TnT (cTnT) or myoglobin.68 However, cTnT has a higher specificity than H-FABP (94% vs. 52%) indicating that elevated H-FABP was found in patients without disease.68 Recent studies have confirmed the utility of using both H-FABP and cTnI for the early diagnosis of MI/ACS.69 Using both markers may also be a valuable rule-out test for patients presenting between 3 and 6 hours after chest pain,69 though H-FABP is not used in the routine evaluation of chest pain.
Ischemia-Modified Albumin
In contrast to the markers described above that detect myocardial tissue damage, the IMA test does not. Instead, it measures changes that occur in albumin in the presence of ischemia, giving it the advantage that it might detect ischemia before damage has occurred to the heart. The free radical formation that occurs during tissue ischemia is believed to modify albumin within minutes of ischemia and lasts for about 6 hours.70, 71, 72, 73 These modifications in albumin alter its ability to bind transition metals, such as cobalt, by its N-terminal domain.72 The amount of IMA is measured by the spectrophotometric determination of albumin’s binding to cobalt.70 While IMA is not specific for cardiac damage, it does have a clinical sensitivity for ACS of 50% to 90%.74,75 Additional studies will be necessary to determine the utility of IMA in the early diagnosis of ACS; its use will complement, not replace, troponin and CK-MB detection.
Novel Markers of Plaque Instability: MPO, CRP, MRP-8/14, and PAPP-A
MPO, CRP, MRP-8/14, and PAPP-A have been studied in international multicenter studies for their utility in the early diagnosis and risk stratification of acute MI in emergency departments in patients with acute chest pain.76 The concentration of all four markers was significantly higher in patients with acute MI compared with patients with other diagnoses.76 But these markers were all inferior to troponins in detecting acute MI.76 MPO, MRP-8/14, and CRP were able to predict all-cause mortality and these provided independent prognostic information apart from clinical risk factors and high-sensitive cTnT; PAPP-A did not.76 Interestingly, these plaque instability markers did not correlate with each other or myocardial cell death determined by troponin.76 The exact mechanisms leading to plaque instability, plaque fissure, and subsequent rupture are not fully understood, but all are expressed in atherosclerotic plaques and contribute to vessel inflammation.77, 78, 79, 80
CASE STUDY 26.3
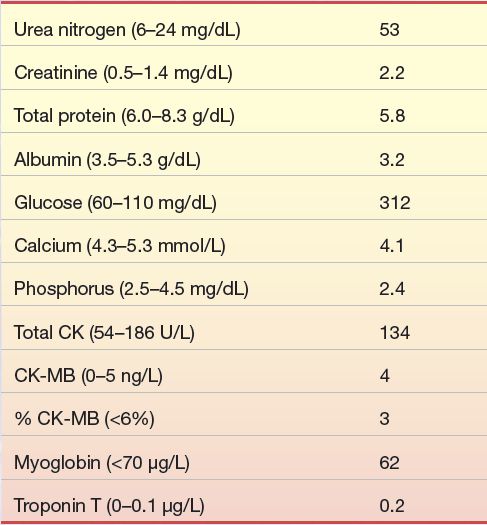
An 83-year-old man with known severe coronary artery disease, diffuse small vessel disease, and significant stenosis distal to a vein graft from previous CABG surgery was admitted when his physician referred him to the hospital after a routine office visit. His symptoms included 3+ pedal edema, jugular vein distention, and heart sound abnormalities. Significant laboratory data obtained on admission were as follows:
Questions
1. Do the symptoms of this patient suggest acute MI?
2. Based on the preceding laboratory data, would this diagnosis be acute MI? Why or why not?
3. Based on the preceding laboratory data, are there other organ system abnormalities present?
4. What are the indicators of these organ system abnormalities?
5. Is there a specific laboratory test that might indicate congestive heart failure in this patient?
CARDIAC INJURY OCCURS IN MANY DISEASE PROCESSES, BEYOND MI
The damage that occurs in the context of MI is due to cardiac muscle cell (myocyte) death. A spectrum of cell death occurs in myocytes, ranging from apoptosis to necrosis to a mixture between the two. Apoptotic cell death is characterized by decreasing cell size, membrane blebbing, nuclear aggregation of chromosomal DNA, and purposeful degradation of DNA. The resulting apoptotic bodies formed from the process of apoptosis are recognized by macrophage, which then remove the debris in an effort to minimize the immune response.81 Apoptosis requires energy, so as ischemia progresses, the apoptotic process stalls and necrosis occurs if energy supply fails to meet the energy demand, which occurs rapidly in the heart. Thus, a rapid clinical response to myocardial ischemia is essential to limiting the extent of necrosis that occurs and is a central tenet of treating MI. The faster cardiac interventions are performed to restore perfusion (along with oxygen) to the heart, the greater the reduction in infarct size. The strategies for reperfusion include percutaneous interventions using intravascular balloons and stents to clear coronary artery blockages and prevent their recurrence, coronary artery bypass grafting (CABG or “bypass surgery”), and the use of chemical thrombolytics, such as tissue plasminogen activator (aka tPA or tenecteplase).
Despite our focus here on the use of cardiac biomarkers in MI, a number of other causes of myocardial injury result in increased biomarker release. Like MI, these disease processes also have myocyte apoptosis and necrosis as part of their pathophysiology, which explains why these biomarkers may be elevated in these patients. Cell death, in the form of apoptosis and/or necrosis, is a key feature of cardiomyopathies due to genetic causes and/or stress-related causes and contributes to the worsening function that occurs with the disease82, 83, 84, 85 (Fig. 26.6). Cell death also occurs in decompensated heart failure and volume overload.86 In the cardiac hypertrophy “pre–heart failure state,” angiotensin II, catecholamine production, and cytokines release play a role in inducing cardiomyocyte apoptosis87, 88, 89 (Fig. 26.6), which can lead to low-level release of cardiac biomarkers. Cardiomyocyte cell death is also induced in myocarditis, whereby viruses and bacteria induce an autoimmune response, which recognizes cardiac-specific proteins as foreign and induces cardiac cells to die.90 Similarly, bacterial toxins in sepsis can cause apoptosis and myocardial depression. Certain drugs are also capable of causing apoptosis and myocardial dysfunction, including doxorubicin and cyclophosphamide91, 92, 93 (Fig. 26.6), as well as alcohol, cocaine, and methamphetamines.
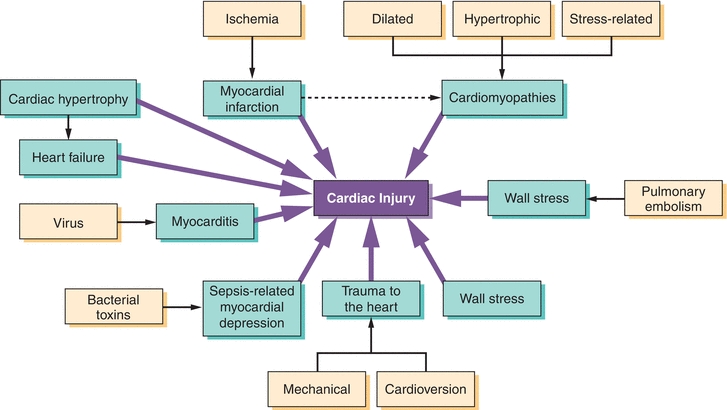
FIGURE 26.6 Common cardiac insults that result in cardiac injury and elevated cardiac biomarkers. (Adapted from McLean A, Huang S. Biomarkers of cardiac injury. In: Vaidya V, Bonventre J, eds. Biomarkers: In Medicine, Drug Discovery, and Environmental Health. New York, NY: John Wiley & Sons, Inc.; 2010:119–155, Ref.85.)
THE LABORATORY WORKUP OF PATIENTS SUSPECTED OF HEART FAILURE AND THE USE OF CARDIAC BIOMARKERS IN HEART FAILURE
Heart failure is a pathological state in which the heart fails to adequately supply the metabolic needs of the body, typically due to a decrease in pumping function. The clinical manifestations of heart failure result largely from the retention of fluid, which is one of the body’s maladaptive responses to decreased cardiac output. The most common symptoms of heart failure are shortness of breath, fatigue, and lower extremity edema.
The ACC/AHA/Heart Failure Society of America and the European Society of Cardiology have recommended specific laboratory and clinical tests for patients with suspected heart failure (see Table 26.4).97, 98, 99 Many of these tests may be familiar as routine laboratory tests; others are more specific to the atherosclerotic process and compensatory cardiac function mechanisms.
TABLE 26.4 American College of Cardiology/American Heart Association/European Society of Cardiology Recommended Laboratory and Clinical Tests for Patients with Suspected Heart Failure

BUN, blood urea nitrogen; AST, aspartate aminotransferase; ALT, alanine aminotransferase; LD, lactate dehydrogenase; BNP, B-type natriuretic peptide; NT-proBNP, N-terminal pro-B–type natriuretic peptide; ECG, electrocardiogram; CV, cardiovascular; LDL, low-density lipoprotein; HDL, high-density lipoprotein; TSH, thyroid-stimulating hormone.
The ACC/AHA recommendations also suggest determining a cTnI or cTnT if the clinical presentation is suggestive of an ACS; elevated troponin levels may also indicate the severity of the heart failure (discussed in the next section).97, 98, 99 A chest x-ray, 2D echocardiographic analysis, and Doppler flow studies to identify ventricular and/or valvular abnormalities are also recommended in evaluating patients with suspected heart failure.100,101 As CHD is the most common cause of heart failure, some evaluation for ischemic burden is indicated. To this end, coronary angiography or exercise stress testing may be used depending on the clinical scenario. Pulmonary function testing is generally not helpful in the diagnosis of heart failure as cardiogenic pulmonary edema and intrinsic lung disease can result in similar patterns of abnormalities.99
THE USE OF NATRIURETIC PEPTIDES AND TROPONINS IN THE DIAGNOSIS AND RISK STRATIFICATION OF HEART FAILURE
Though shortness of breath is the most common presentation of heart failure, it is a relatively nonspecific symptom. Measurement of circulating B-type natriuretic peptide (BNP), or its precursor N-terminal pro-B–type natriuretic peptide (NT-proBNP), can be particularly helpful in distinguishing cardiac from noncardiac causes of dyspnea and is widely used in emergency departments and other clinical settings. 97, 98, 99,102, 103, 104, 105
Natriuretic peptides are secreted from the heart in response to increased pressure and volume load. They play an important role in reducing intravascular volume by promoting natriuresis, diuresis, and vasodilation and inhibiting sympathetic nervous system signaling.106,107 The proBNP is released by myocardial cells in response to increased volume, increased pressure, and cardiac hypertrophy; this precursor is cleaved by the protease enzyme furin into the active BNP and the inactive NT-proBNP. Both NT-proBNP and BNP are elevated in patients with ventricular dysfunction and strongly predict morbidity and mortality in patients with heart failure.108, 109, 110, 111 Multiple clinical studies have assessed the utility of both NT-proBNP and BNP for ruling out heart failure as a cause of dyspnea (shortness of breath) in the acute clinical setting.111 Meta-analysis of these studies have found that the pooled estimates of sensitivity and specificity are equivalent for NT-proBNP and BNP. However, the optimum cutoff value for each peptide remains difficult to determine across all populations.111
Vasodilation and natriuresis are beneficial in the setting of heart failure, and thus, BNP was pursued as a target for drug development. Scios (later Johnson & Johnson) developed recombinant BNP as nesiritide. It is the first in a new class of therapies designed to treat heart failure, acting as a neurohormonal suppressor just as endogenous BNP.112 The importance of mentioning that recombinant BNP is used therapeutically is that it can be picked up by laboratory tests for BNP, but not NT-proBNP. Therefore, there are clinical situations wherein NT-proBNP may be the more appropriate test to use to follow heart failure patients.
Cardiac Troponins
While used primarily for the diagnosis of myocardial injury caused by ischemia, elevations of cTnT and cTnI levels were recognized in heart failure patients more than a decade ago.113, 114, 115 The precise reason for elevated troponins in the non-ACS setting of heart failure is not clear. Ongoing cell death including apoptosis and necrosis may be one of the reasons, occurring as a result of increased myocardial wall stress116 resulting in subendocardial ischemia due to increased myocardial oxygen demand. Diminished cardiac perfusion and oxygen delivery to the heart itself and impaired renal clearance of troponins may also contribute.116
While the detection of troponins in heart failure is not diagnostic, it has been reported to add prognostic value. In acute heart failure patients without ACS, elevations in cTnI occur more often than cTnT elevations, although increases in either were related to increased mortality.116 Other studies have described a 2.6-fold increased risk of in-hospital mortality for heart failure patients with elevated troponin levels at the time of admission.117 Elevated cardiac troponin has also been associated with lower systolic blood pressure and lower left ventricular ejection fraction at the time of admission,117 both of which are markers for worse outcome. In outpatients with more severe chronic heart failure (New York Heart Failure Class III or IV), the presence of elevated cardiac troponin was also associated with lower ejection fraction and deteriorating clinical course.115 Troponin levels were one of the strongest predictors of mortality, particularly if used in conjunction with BNP levels.118 In general, concomitant elevations in multiple markers (cardiac troponin, high-sensitivity CRP [hsCRP], along with NT-proBNP) are associated with escalating risks of adverse events.119 Few studies, however, have investigated how troponin levels may be helpful in the initial diagnosis of heart failure, so their current role in heart failure patients is limited to risk stratification.
MARKERS OF CHD RISK
C-Reactive Protein
Inflammation plays an important role in the development and progression of atherosclerosis and CHD. CRP is an acute marker of inflammation that is currently used clinically in the evaluation of CVD risk. CRP is a pentameric protein consisting of five identical subunits that bind to specific ligands, such as LDL cholesterol, in a calcium-dependent manner. CRP is normally present in human plasma at levels less than 10 mg/L, but its rapid synthesis in the liver after stimulus from a variety of inflammatory cytokines may increase plasma levels by 1,000-fold, thus serving as a sensitive biomarker of systemic inflammation.120 Because atherosclerosis and CHD derive largely from an inflammatory etiology, CRP has long been targeted as a biomarker for CHD, but it has recently gained widest acceptance as a marker of CHD risk.
CASE STUDY 26.4
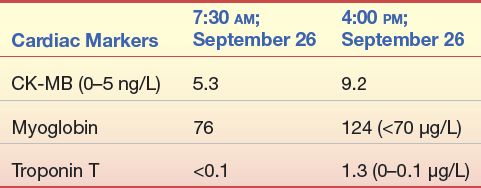
A 68-year-old man presented to the emergency department with sudden onset of chest pain, left arm pain, dyspnea, and weakness while away from home on a business trip. His prior medical history is not available, but he admits to being a 2-pack per day smoker for longer than 20 years.
Cardiac markers were performed at admission and 8 hours postadmission with the following results:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


