Adapted from Competence Assurance, ASMT. Enzymology, An Educational Program. Bethesda, MD: RMI Corporation; 1980.
Table 13.1 also lists common and standard abbreviations for commonly analyzed enzymes. Without IUB recommendation, capital letters have been used as a convenience to identify enzymes. The common abbreviations, sometimes developed from previously accepted names for the enzymes, were used until the standard abbreviations listed in the table were developed.2,3 These standard abbreviations are used in the United States and are used later in this chapter to indicate specific enzymes.
ENZYME KINETICS
Catalytic Mechanism of Enzymes
There are some chemical reactions that may occur at a slow rate if there is not enough kinetic energy to drive the reaction to the formation of products (uncatalyzed reaction). Other chemical reactions may occur spontaneously if the free energy or available kinetic energy is higher for the reactants than for the products. The reaction then proceeds toward the lower energy if a sufficient number of the reactant molecules possess enough excess energy to break their chemical bonds and collide to form new bonds. The excess energy, called activation energy, is the energy required to raise all molecules in 1 mole of a compound at a certain temperature to the transition state at the peak of the energy barrier. At the transition state, each molecule is equally likely to either participate in product formation or remain an unreacted molecule. Reactants possessing enough energy to overcome the energy barrier participate in product formation.
One way to provide more energy for a reaction is to increase the temperature, which will increase intermolecular collisions; however, this does not normally occur physiologically. Enzymes catalyze physiologic reactions by lowering the activation energy level that the reactants (substrates) must reach for the reaction to occur (Fig. 13.1). The reaction may then occur more readily to a state of equilibrium in which there is no net forward or reverse reaction, even though the equilibrium constant of the reaction is not altered. The extent to which the reaction progresses depends on the number of substrate molecules that pass the energy barrier.
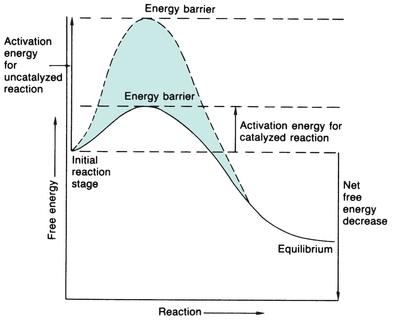
FIGURE 13.1 Energy versus progression of reaction, indicating the energy barrier that the substrate must surpass to react with and without enzyme catalysis. The enzyme considerably reduces the free energy needed to activate the reaction.
The general relationship among enzyme, substrate, and product may be represented as follows:
 (Eq. 13-1)
(Eq. 13-1) where E is enzyme, S is substrate, ES is enzyme–substrate complex, and P is product.
The ES complex is a physical binding of a substrate to the active site of an enzyme. The structural arrangement of amino acid residues within the enzyme makes the three-dimensional active site available. At times, the binding of ligand drives a rearrangement to make the active site. The transition state for the ES complex has a lower energy of activation than the transition state of S alone, so that the reaction proceeds after the complex is formed. An actual reaction may involve several substrates and products.
Different enzymes are specific to substrates in different extents or respects. Certain enzymes exhibit absolute specificity, meaning that the enzyme combines with only one substrate and catalyzes only the one corresponding reaction. Other enzymes are group specific because they combine with all substrates containing a particular chemical group, such as a phosphate ester. Still other enzymes are specific to chemical bonds and thereby exhibit bond specificity.
Stereoisomeric specificity refers to enzymes that predominantly combine with only one optical isomer of a certain compound. In addition, an enzyme may bind more than one molecule of substrate, and this may occur in a cooperative fashion. Binding of one substrate molecule, therefore, may facilitate binding of additional substrate molecules.
Factors That Influence Enzymatic Reactions
Substrate Concentration
The rate at which an enzymatic reaction proceeds and whether the forward or reverse reaction occurs depend on several reaction conditions. One major influence on enzymatic reactions is substrate concentration. In 1913, Michaelis and Menten hypothesized the role of substrate concentration in formation of the enzyme–substrate (ES) complex. According to their hypothesis, represented in Figure 13.2, the substrate readily binds to free enzyme at a low substrate concentration. The reaction rate steadily increases when more substrate is added, with the amount of enzyme exceeding the amount of substrate. When this occurs, the reaction is following first-order kinetics because the reaction rate is directly proportional to substrate concentration. Eventually, when the substrate concentration is high enough to saturate all available enzyme, the reaction velocity has reached its maximum. When product is formed, the resultant free enzyme immediately combines with excess free substrate. The reaction is in zero-order kinetics, and the reaction rate depends only on enzyme concentration.
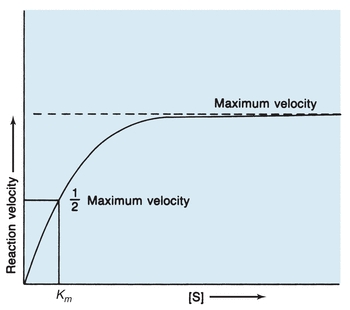
FIGURE 13.2 Michaelis-Menten curve of velocity versus substrate concentration for enzymatic reaction. Km is the substrate concentration at which the reaction velocity is half of the maximum level.
The Michaelis-Menten constant (Km), derived from the theory of Michaelis and Menten, is a constant for a specific enzyme and substrate under defined reaction conditions and is an expression of the relationship between the velocity of an enzymatic reaction and substrate concentration. The assumptions are made that equilibrium among E, S, ES, and P is established rapidly and that the E + P → ES reaction is negligible. The rate-limiting step is the formation of product and enzyme from the ES complex. Then, maximum velocity is fixed, and the reaction rate is a function of only the enzyme concentration. As designated in Figure 13.2, Km is specifically the substrate concentration at which the enzyme yields half the possible maximum velocity. Therefore, Km indicates the amount of substrate needed for a particular enzymatic reaction.
The Michaelis-Menten hypothesis of the relationship between reaction velocity and substrate concentration can be represented mathematically as follows:
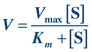 (Eq. 13-2)
(Eq. 13-2) where V is measured velocity of reaction, Vmax is maximum velocity, [S] is substrate concentration, and Km is Michaelis-Menten constant of enzyme for specific substrate.
Theoretically, Vmax and then Km could be determined from the plot in Figure 13.2. However, Vmax is difficult to determine from the hyperbolic plot and often not actually achieved in enzymatic reactions because enzymes may not function optimally in the presence of excessive substrate. A more accurate and convenient determination of Vmax and Km may be made through a Lineweaver-Burk plot, a double-reciprocal plot of the Michaelis-Menten constant, which yields a straight line (Fig. 13.3). The reciprocal is taken of both the substrate concentration and the velocity of an enzymatic reaction. The equation becomes
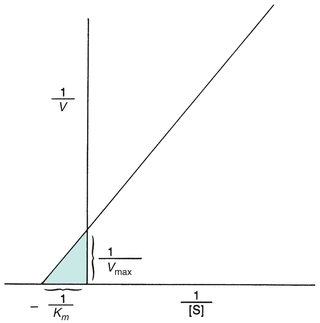
FIGURE 13.3 Lineweaver-Burk transformation of Michaelis-Menten curve. Vmax is the reciprocal of the x-intercept of the straight line. Km is the negative reciprocal of the x-intercept of the same line.
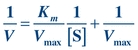 (Eq. 13-3)
(Eq. 13-3) Enzyme Concentration
Because enzymes catalyze physiologic reactions, the enzyme concentration affects the rate of the catalyzed reaction. As long as the substrate concentration exceeds the enzyme concentration, the velocity of the reaction is proportional to the enzyme concentration. The higher the enzyme level, the faster the reaction will proceed because more enzyme is present to bind with the substrate.
pH
Enzymes are proteins that carry net molecular charges. Changes in pH may denature an enzyme or influence its ionic state, resulting in structural changes or a change in the charge on an amino acid residue in the active site. Hence, each enzyme operates within a specific pH range and maximally at a specific pH. Most physiologic enzymatic reactions occur in the pH range of 7.0 to 8.0, but some enzymes are active in wider pH ranges than others. In the laboratory, the pH for a reaction is carefully controlled at the optimal pH by means of appropriate buffer solutions.
Temperature
Increasing temperature usually increases the rate of a chemical reaction by increasing the movement of molecules, the rate at which intermolecular collisions occur, and the energy available for the reaction. This is the case with enzymatic reactions until the temperature is high enough to denature the protein composition of the enzyme. For each 10 degree increase in temperature, the rate of the reaction will approximately double until, of course, the protein is denatured.
Each enzyme functions optimally at a particular temperature, which is influenced by other reaction variables, especially the total time for the reaction. The optimal temperature is usually close to that of the physiologic environment of the enzyme; however, some denaturation may occur at the human physiologic temperature of 37°C. The rate of denaturation increases as the temperature increases and is usually significant at 40°C to 50°C.
Because low temperatures render enzymes reversibly inactive, many serum or plasma specimens for enzyme measurement are refrigerated or frozen to prevent activity loss until analysis. Storage procedures may vary from enzyme to enzyme because of individual stability characteristics. Repeated freezing and thawing, however, tends to denature protein and should be avoided.
Because of their temperature sensitivity, enzymes should be analyzed under strictly controlled temperature conditions. Incubation temperatures should be accurate within ±0.1°C. Laboratories usually attempt to establish an analysis temperature for routine enzyme measurement of 25°C, 30°C, or 37°C. Attempts to establish a universal temperature for enzyme analysis have been unsuccessful and, therefore, reference ranges for enzyme levels may vary significantly among laboratories. In the United States, however, 37°C is most commonly used.
Cofactors
Cofactors are nonprotein entities that must bind to particular enzymes before a reaction occurs. Common activators (inorganic cofactors) are metallic (Ca2+, Fe2+, Mg2+, Mn2+, Zn2+, and K+) and nonmetallic (Br− and Cl−). The activator may be essential for the reaction or may only enhance the reaction rate in proportion with concentration to the point at which the excess activator begins to inhibit the reaction. Activators function by alternating the spatial configuration of the enzyme for proper substrate binding, linking substrate to the enzyme or coenzyme, or undergoing oxidation or reduction.
Some common coenzymes (organic cofactors) are nucleotide phosphates and vitamins. Coenzymes serve as second substrates for enzymatic reactions. When bound tightly to the enzyme, coenzymes are called prosthetic groups. For example, NAD as a cofactor may be reduced to nicotinamide adenine dinucleotide phosphate (NADP) in a reaction in which the primary substrate is oxidized. Increasing coenzyme concentration will increase the velocity of an enzymatic reaction in a manner synonymous with increasing substrate concentration. When quantitating an enzyme that requires a particular cofactor, that cofactor should always be provided in excess so that the extent of the reaction does not depend on the concentration of the cofactor.
Inhibitors
Enzymatic reactions may not progress normally if a particular substance, an inhibitor, interferes with the reaction. Competitive inhibitors physically bind to the active site of an enzyme and compete with the substrate for the active site. With a substrate concentration significantly higher than the concentration of the inhibitor, the inhibition is reversible because the substrate is more likely than the inhibitor to bind the active site and the enzyme has not been destroyed.
A noncompetitive inhibitor binds an enzyme at a place other than the active site and may be reversible in the respect that some naturally present metabolic substances combine reversibly with certain enzymes. Noncompetitive inhibition also may be irreversible if the inhibitor destroys part of the enzyme involved in catalytic activity. Because the inhibitor binds the enzyme independently from the substrate, increasing substrate concentration does not reverse the inhibition.
Uncompetitive inhibition is another kind of inhibition in which the inhibitor binds to the ES complex—increasing substrate concentration results in more ES complexes to which the inhibitor binds and, thereby, increases the inhibition. The enzyme–substrate–inhibitor complex does not yield product. Lastly, a mixed inhibitor has the ability to bind to either the E or ES complex at a different site from the substrate active site.
Each of the four types of inhibition are unique with respect to effects on the Vmax and Km of enzymatic reactions (Fig. 13.4). In competitive inhibition, the effect of the inhibitor can be counteracted by adding excess substrate to bind the enzyme. The amount of the inhibitor is then negligible by comparison, and the reaction will proceed at a slower rate but to the same maximum velocity as an uninhibited reaction. The Km is a constant for each enzyme and cannot be altered. However, because the amount of substrate needed to achieve a particular velocity is higher in the presence of a competing inhibitor, the Km appears to increase when exhibiting the effect of the inhibitor.
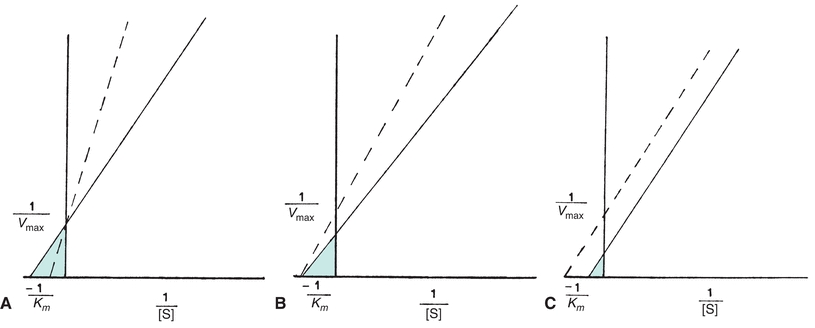
FIGURE 13.4 Normal Lineweaver-Burk plot (solid line) compared with each type of enzyme inhibition (dotted line). (A) Competitive inhibition Vmax unaltered; Km appears increased. (B) Noncompetitive inhibition Vmax decreased; Km unchanged. (C) Uncompetitive inhibition Vmax decreased; Km appears decreased.
The substrate and inhibitor, commonly a metallic ion, may bind an enzyme simultaneously in noncompetitive inhibition. The inhibitor may inactivate either an ES complex or just the enzyme by causing structural changes in the enzyme. Even if the inhibitor binds reversibly and does not inactivate the enzyme, the presence of the inhibitor when it is bound to the enzyme slows the rate of the reaction. Thus, for noncompetitive inhibition, the maximum reaction velocity cannot be achieved. Increasing substrate levels has no influence on the binding of a noncompetitive inhibitor, so the Km is unchanged.
Because uncompetitive inhibition requires the formation of an ES complex, increasing substrate concentration increases inhibition. Therefore, maximum velocity equal to that of an uninhibited reaction cannot be achieved, and the Km appears to be decreased.
In addition, mixed inhibitors have the potential to interfere with substrate binding and enzyme catalysis. Consequently, the apparent Vmax may decrease, and the apparent Km may decrease or increase.
Measurement of Enzyme Activity
Because enzymes are usually present in very small quantities in biologic fluids and often difficult to isolate from similar compounds, a convenient method of enzyme quantitation is measurement of catalytic activity. Activity is then related to concentration. Common methods might photometrically measure an increase in product concentration, a decrease in substrate concentration, a decrease in coenzyme concentration, or an increase in the concentration of an altered coenzyme.
If the amount of substrate and any coenzyme is in excess in an enzymatic reaction, the amount of substrate or coenzyme used, or product or altered coenzyme formed, will depend only on the amount of enzyme present to catalyze the reaction. Enzyme concentrations, therefore, are always performed in zero-order kinetics, with the substrate in sufficient excess to ensure that no more than 20% of the available substrate is converted to product. Any coenzymes also must be in excess. NADH is a coenzyme frequently measured in the laboratory. NADH absorbs light at 340 nm, whereas NAD does not, and a change in absorbance at 340 nm is easily measured.
In specific laboratory methodologies, substances other than substrate or coenzyme are necessary and must be present in excess. NAD or NADH is often convenient as a reagent for a coupled enzyme assay when neither NAD nor NADH is a coenzyme for the reaction. In other coupled enzyme assays, more than one enzyme is added in excess as a reagent and multiple reactions are catalyzed. After the enzyme under analysis catalyzes its specific reaction, a product of that reaction becomes the substrate on which an intermediate auxiliary enzyme acts. A product of the intermediate reaction becomes the substrate for the final reaction, which is catalyzed by an indicator enzyme and commonly involves the conversion of NAD to NADH or vice versa.
When performing an enzyme quantitation in zero-order kinetics, inhibitors must not be present and other variables that may influence the rate of the reaction must be carefully controlled. A constant pH should be maintained by means of an appropriate buffer solution. The temperature should be constant within ±0.1°C throughout the assay at a temperature at which the enzyme is active (usually, 25°C, 30°C, or 37°C).
During the progress of the reaction, the period for the analysis also must be carefully selected. When the enzyme is initially introduced to the reactants and the excess substrate is steadily combining with available enzyme, the reaction rate rises. After the enzyme is saturated, the rates of product formation, release of enzyme, and recombination with more substrate proceed linearly. After a time, usually 6 to 8 minutes after reaction initiation, the reaction rate decreases as the substrate is depleted, the reverse reaction is occurring appreciably, and the product begins to inhibit the reaction. Hence, enzyme quantitations must be performed during the linear phase of the reaction.
One of two general methods may be used to measure the extent of an enzymatic reaction: (1) fixed-time and (2) continuous-monitoring or kinetic assay. In the fixed-time method, the reactants are combined, the reaction proceeds for a designated time, the reaction is stopped (usually by inactivating the enzyme with a weak acid), and a measurement is made of the amount of reaction that has occurred. The reaction is assumed to be linear over the reaction time; the larger the reaction, the more enzyme is present.
In continuous-monitoring or kinetic assays, multiple measurements, usually of absorbance change, are made during the reaction, either at specific time intervals (usually every 30 or 60 seconds) or continuously by a continuous-recording spectrophotometer. These assays are advantageous over fixed-time methods because the linearity of the reaction may be more adequately verified. If absorbance is measured at intervals, several data points are necessary to increase the accuracy of linearity assessment. Continuous measurements are preferred because any deviation from linearity is readily observable.
The most common cause of deviation from linearity occurs when the enzyme is so elevated that all substrate is used early in the reaction time. For the remainder of the reaction, the rate change is minimal, with the implication that the coenzyme concentration is very low. With continuous monitoring, the laboratorian may observe a sudden decrease in the reaction rate (deviation from zero-order kinetics) of a particular determination and may repeat the determination using less patient sample. The decrease in the amount of patient sample operates as a dilution, and the answer obtained may be multiplied by the dilution factor to obtain the final answer. The sample itself is not diluted so that the diluent cannot interfere with the reaction. (Sample dilution with saline may be necessary to minimize negative effects in analysis caused by hemolysis or lipemia.) Enzyme activity measurements may not be accurate if storage conditions compromise integrity of the protein, if enzyme inhibitors are present, or if necessary cofactors are not present.
Calculation of Enzyme Activity
When enzymes are quantified relative to their activity rather than a direct measurement of concentration, the units used to report enzyme levels are activity units. The definition for the activity unit must consider variables that may alter results (e.g., pH, temperature, substrate). Historically, specific method developers frequently established their own units for reporting results and often named the units after themselves (i.e., Bodansky and King units). To standardize the system of reporting quantitative results, the EC defined the international unit (IU) as the amount of enzyme that will catalyze the reaction of 1 μmol of substrate per minute under specified conditions of temperature, pH, substrates, and activators. Since specified conditions may vary among laboratories, reference values are still often laboratory specific. Enzyme concentration is usually expressed in units per liter (IU/L). The unit of enzyme activity recognized by the International System of Units (Système International d’Unités [SI]) is the katal (mol/s). The mole is the unit for substrate concentration, and the unit of time is the second. Enzyme concentration is then expressed as katals per liter (kat/L) (1.0 IU = 17 nkat).
When enzymes are quantitated by measuring the increase or decrease of NADH at 340 nm, the molar absorptivity (6.22 × 103 mol/L) of NADH is used to calculate enzyme activity.
Measurement of Enzyme Mass
Immunoassay methodologies that quantify enzyme concentration by mass are also available and are routinely used for quantification of some enzymes, such as creatine kinase (CK)-MB. Immunoassays may overestimate active enzyme as a result of possible cross-reactivity with inactive enzymes, such as zymogens, inactive isoenzymes, macroenzymes, or partially digested enzyme. The relationship between enzyme activity and enzyme quantity is generally linear but should be determined for each enzyme. Enzymes may also be determined and quantified by electrophoretic techniques, which provide resolution of isoenzymes and isoforms.
Ensuring the accuracy of enzyme measurements has long been a concern of laboratorians. The Clinical Laboratory Improvement Amendment of 1988 (CLIA ’88) has established guidelines for quality control and proficiency testing for all laboratories. Problems with quality control materials for enzyme testing have been a significant issue. Differences between clinical specimens and control sera include species of origin of the enzyme, integrity of the molecular species, isoenzyme forms, matrix of the solution, addition of preservatives, and lyophilization processes. Many studies have been conducted to ensure accurate enzyme measurements and good quality control materials.4
Enzymes as Reagents
Enzymes may be used as reagents to measure many nonenzymatic constituents in serum. For example, glucose, cholesterol, and uric acid are frequently quantified by means of enzymatic reactions, which measure the concentration of the analyte due to the specificity of the enzyme. Enzymes are also used as reagents for methods to quantify analytes that are substrates for the corresponding enzyme. One example, lactate dehydrogenase (LDH), may be a reagent when lactate or pyruvate concentrations are evaluated. For such methods, the enzyme is added in excess in a quantity sufficient to provide a complete reaction in a short period.
Immobilized enzymes are chemically bonded to adsorbents, such as agarose or certain types of cellulose, by azide groups, diazo, and triazine. The enzymes act as recoverable reagents. When substrate is passed through the preparation, the product is retrieved and analyzed, and the enzyme is present and free to react with more substrate. Immobilized enzymes are convenient for batch analyses and are more stable than enzymes in a solution. Enzymes are also commonly used as reagents in competitive and noncompetitive immunoassays, such as those used to measure human immunodeficiency virus (HIV) antibodies, therapeutic drugs, and cancer antigens. Commonly used enzymes include horseradish peroxidase, alkaline phosphatase (ALP), glucose-6-phosphate dehydrogenase, and β-galactosidase. The enzyme in these assays functions as an indicator that reflects either the presence or absence of the analyte.
ENZYMES OF CLINICAL SIGNIFICANCE
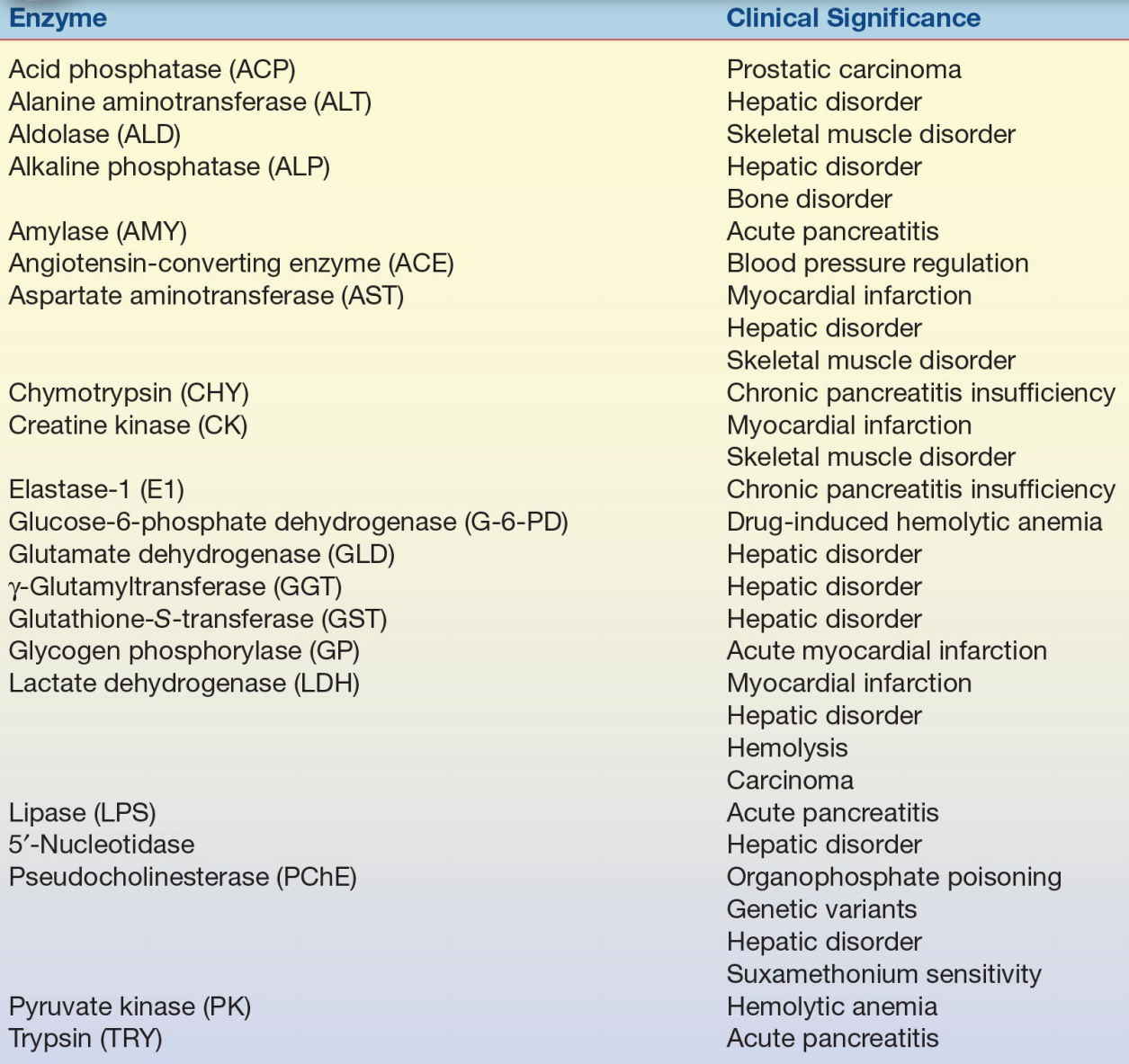
Table 13.2 lists the commonly analyzed enzymes, including their systematic names and clinical significance. Each enzyme is discussed in this chapter with respect to tissue source, diagnostic significance, assay method, source of error, and reference range.
TABLE 13.2 Major Enzymes of Clinical Significance
CASE STUDY 13.1
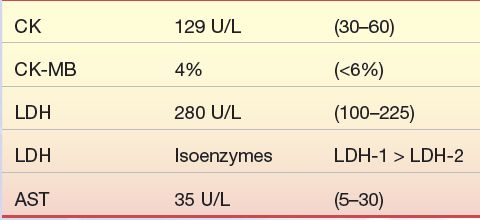
A 57-year-old moderately overweight Caucasian male visits his family physician with a symptom of “indigestion” of 5 days’ duration. He has also had bouts of sweating, malaise, and headache. His blood pressure is 140/105 mm Hg; his family history includes a father with diabetes who died at age 62 of AMI secondary to diabetes mellitus. An electrocardiogram revealed changes from one performed 6 months earlier. The results of the patient’s blood work are as follows:
Questions
1. Can a diagnosis of AMI be ruled out in this patient?
2. What further cardiac markers should be run on this patient?
3. Should this patient be admitted to the hospital?
Creatine Kinase
CK is an enzyme with a molecular weight of approximately 82,000 that is generally associated with ATP regeneration in contractile or transport systems. Its predominant physiologic function occurs in muscle cells, where it is involved in the storage of high-energy creatine phosphate. Every contraction cycle of muscle results in creatine phosphate use, with the production of ATP. This results in relatively constant levels of muscle ATP. The reversible reaction catalyzed by CK is shown in Equation 13-4.
 (Eq. 13-4)
(Eq. 13-4) Tissue Source
CK is widely distributed in tissue, with highest activities found in skeletal muscle, heart muscle, and brain tissue. CK is present in much smaller quantities in other tissue sources, including the bladder, placenta, gastrointestinal tract, thyroid, uterus, kidney, lung, prostate, spleen, liver, and pancreas.
Diagnostic Significance
Due to the high concentrations of CK in muscle tissue, CK levels are frequently elevated in disorders of cardiac and skeletal muscle (myocardial infarction [MI], rhabdomyolysis, and muscular dystrophy). The CK level is considered a sensitive indicator of acute myocardial infarction (AMI) and muscular dystrophy, particularly the Duchenne type. Extreme elevations of CK occur in Duchenne-type muscular dystrophy, with values reaching 50 to 100 times the upper limit of normal (ULN). Although total CK levels are sensitive indicators of these disorders, they are not entirely specific indicators inasmuch as CK elevation is found in various other abnormal cardiac and skeletal muscle conditions. Levels of CK also vary with muscle mass and, therefore, may depend on gender, race, degree of physical conditioning, and age.
Elevated CK levels are also occasionally seen in central nervous system disorders such as cerebrovascular accident, seizures, nerve degeneration, and central nervous system shock. Damage to the blood–brain barrier must occur to allow enzyme release to the peripheral circulation.
Other pathophysiologic conditions in which elevated CK levels occur are hypothyroidism, malignant hyperpyrexia, and Reye’s syndrome. Table 13.3 lists the major disorders associated with abnormal CK levels. Serum CK levels and CK/progesterone ratio have been useful in the diagnosis of ectopic pregnancies.5 Total serum CK levels have also been used as an early diagnostic tool to identify patients with Vibrio vulnificus infections.6
TABLE 13.3 Creatine Kinase Isoenzymes—Tissue Localization and Sources of Elevation
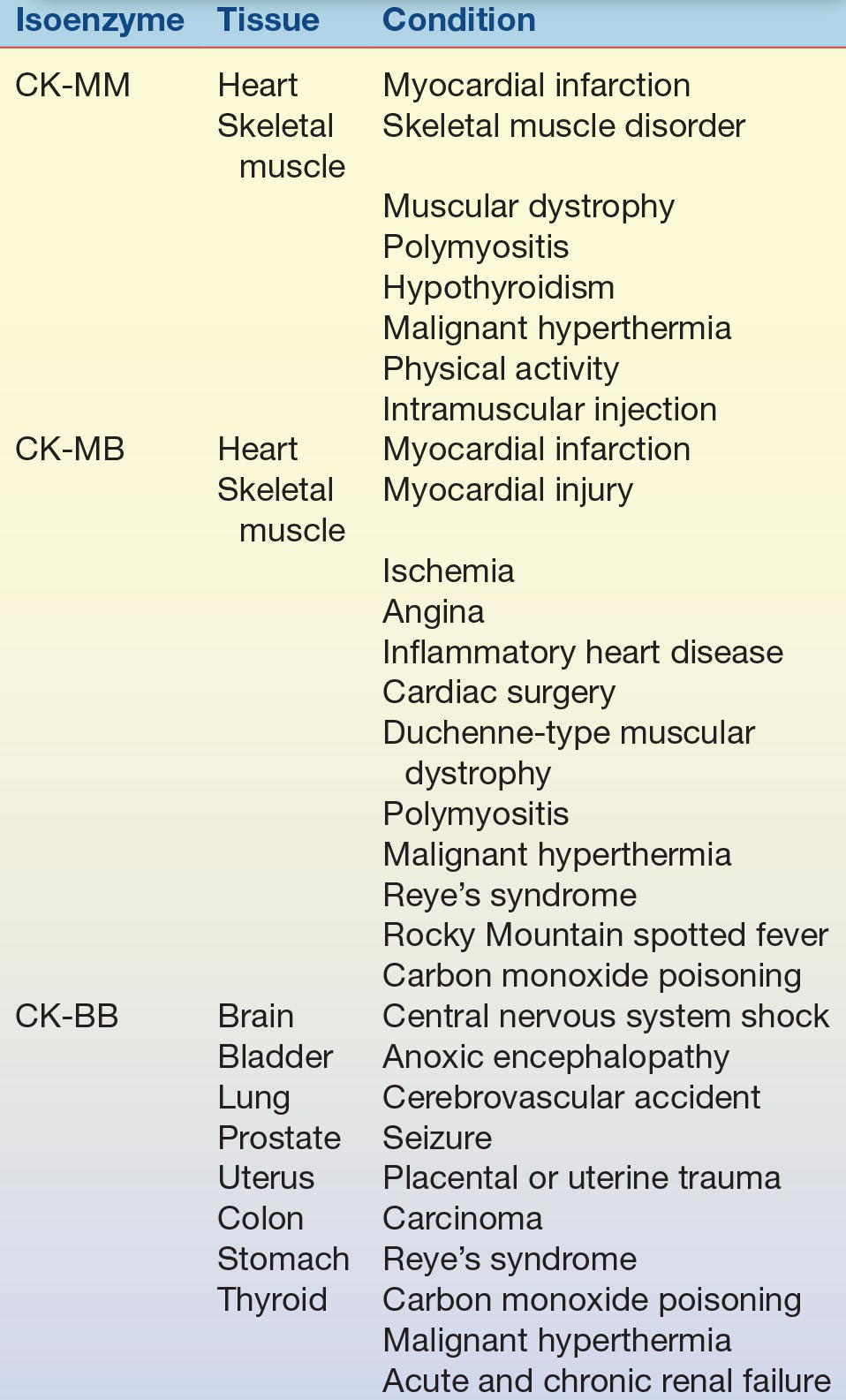
Because enzyme elevation is found in numerous disorders, the separation of total CK into its various isoenzyme fractions is considered a more specific indicator of various disorders than total levels. Typically, the clinical relevance of CK activity depends more on isoenzyme fractionation than on total levels.
CK occurs as a dimer consisting of two subunits that can be separated readily into three distinct molecular forms. The three isoenzymes have been designated as CK-BB (brain type), CK-MB (hybrid type), and CK-MM (muscle type). On electrophoretic separation, CK-BB will migrate fastest toward the anode and is therefore called CK-1. CK-BB is followed by CK-MB (CK-2) and, finally, by CK-MM (CK-3), exhibiting the slowest mobility (Fig. 13.5). Table 13.3 indicates the tissue localization of the isoenzymes and the major conditions associated with elevated levels. Separation of CK isoforms may also by visualized by high-voltage electrophoretic separation. Isoforms occur following cleavage of the carboxyl-terminal amino acid from the M subunit by serum carboxypeptidase N. Three isoforms have been described for CK-MM and two isoforms for CK-MB; the clinical significance is not well established.
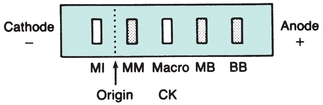
FIGURE 13.5 Electrophoretic migration pattern of normal and atypical CK isoenzymes.
The major isoenzyme in the sera of healthy people is the MM form. Values for the MB isoenzyme range from undetectable to trace (<6% of total CK). It also appears that CK-BB is present in small quantities in the sera of healthy people; however, the presence of CK-BB in serum depends on the method of detection. Most techniques cannot detect CK-BB in normal serum.
CK-MM is the major isoenzyme fraction found in striated muscle and normal serum. Skeletal muscle contains almost entirely CK-MM, with a small amount of CK-MB. The majority of CK activity in heart muscle is also attributed to CK-MM, with approximately 20% a result of CK-MB.7 Normal serum consists of approximately 94% to 100% CK-MM. Injury to both cardiac and skeletal muscle accounts for the majority of cases of CK-MM elevations (Table 13.3). Hypothyroidism results in CK-MM elevations because of the involvement of muscle tissue (increased membrane permeability), the effect of thyroid hormone on enzyme activity, and, possibly, the slower clearance of CK as a result of slower metabolism.
Mild to strenuous activity may contribute to elevated CK levels, as may intramuscular injections. In physical activity, the extent of elevation is variable. However, the degree of exercise in relation to the exercise capacity of the individual is the most important factor in determining the degree of elevation.8 Patients who are physically well conditioned show lesser degrees of elevation than do patients who are less conditioned. Levels may be elevated for as long as 48 hours following exercise. CK elevations are generally less than five times ULN following intramuscular injections and usually not apparent after 48 hours, although elevations may persist for 1 week. The predominant isoenzyme is CK-MM.
The quantity of CK-BB in the tissue (Table 13.3) is usually small. The small quantity, coupled with its relatively short half-life (1 to 5 hours), results in CK-BB activities that are generally low and transient and not usually measurable when tissue damage occurs. Highest concentrations are found in the central nervous system, the gastrointestinal tract, and the uterus during pregnancy. Although brain tissue has high concentrations of CK, serum rarely contains CK-BB of brain origin. Because of its molecular size (80,000), its passage across the blood–brain barrier is hindered. However, when extensive damage to the brain has occurred, significant amounts of CK-BB can sometimes be detected in the serum. It has been observed that CK-BB may be significantly elevated in patients with carcinoma of various organs. It has been found in association with untreated prostatic carcinoma and other adenocarcinomas. These findings indicate that CK-BB may be a useful tumor-associated marker.9 The most common causes of CK-BB elevations are central nervous system damage, tumors, childbirth, and the presence of macro-CK, an enzyme–immunoglobulin complex. In most of these cases, the CK-BB level is greater than 5 U/L, usually in the range of 10 to 50 U/L. Other conditions listed in Table 13.3 usually show CK-BB activity below 10 U/L.10
The value of CK isoenzyme separation can be found principally in detection of myocardial damage. Cardiac tissue contains significant quantities of CK-MB, approximately 20% of all CK-MB. Whereas CK-MB is found in small quantities in other tissue, myocardium is essentially the only tissue from which CK-MB enters the serum in significant quantities. Demonstration of elevated levels of CK-MB, greater than or equal to 6% of the total CK, is considered a good indicator of myocardial damage, particularly AMI. Other nonenzyme proteins, called troponins, have been found to be even more specific and may elevate in the absence of CK-MB elevations. Following MI, the CK-MB levels begin to rise within 4 to 8 hours, peak at 12 to 24 hours, and return to normal levels within 48 to 72 hours. This time frame must be considered when interpreting CK-MB levels.
CK-MB activity has been observed in other cardiac disorders (Table 13.3). Therefore, increased quantities are not entirely specific for AMI but probably reflect some degree of ischemic heart damage. Consequently, troponin is the preferred marker to detect MI because it is the most sensitive and specific for myocardial damage. The specificity of CK-MB levels in the diagnosis of AMI can be increased if interpreted in conjunction with LDH isoenzymes and/or troponins and if measured sequentially over a 48-hour period to detect the typical rise and fall of enzyme activity seen in AMI (Fig. 13.6). The MB isoenzyme also has been detected in the sera of patients with noncardiac disorders. CK-MB levels found in these conditions probably represent leakage from skeletal muscle, although in Duchenne-type muscular dystrophy, there may be some cardiac involvement as well. CK-MB levels in Reye’s syndrome also may reflect myocardial damage. Despite the findings of CK-MB levels in disorders other than MI, its presence still remains a significant indicator of AMI.11 The typical time course of CK-MB elevation following AMI is not found in other conditions.
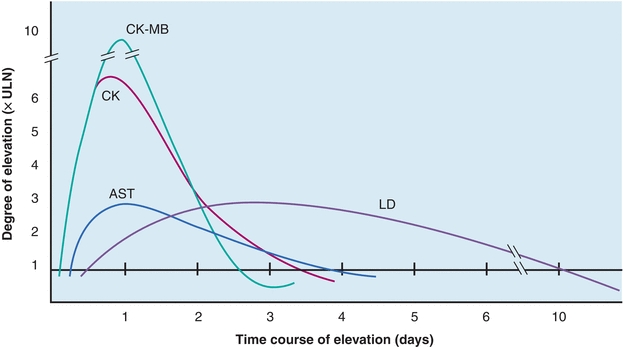
FIGURE 13.6 Time–activity curves of enzymes in myocardial infarction for AST, CK, CK-MB, and LDH. CK, specifically the MB fraction, increases initially, followed by AST and LDH. LDH is elevated the longest. All enzymes usually return to normal within 10 days.
Nonenzyme proteins (troponin I and troponin T) have been used as a more sensitive and specific marker of myocardial damage. These proteins are released into the bloodstream earlier and persist longer than CK and its isoenzyme CK-MB. More information on these protein markers of AMI can be found in Chapters 10 and 26.
Numerous reports have been made describing the appearance of unusual CK isoenzyme bands displaying electrophoretic properties that differ from the three major isoenzyme fractions12, 13, 14, 15, 16 (Fig. 13.5). These atypical forms are generally of two types and are referred to as macro-CK and mitochondrial CK.
Macro-CK appears to migrate to a position midway between CK-MM and CK-MB. This type of macro-CK largely comprises CK-BB complexed with immunoglobulin. In most instances, the associated immunoglobulin is IgG, although a complex with IgA also has been described. The term macro-CK has also been used to describe complexes of lipoproteins with CK-MM. The incidence of macro-CK in sera ranges from 0.8% to 1.6%. Currently, no specific disorder is associated with its presence, although it appears to be age and sex related, occurring most frequently in women older than age 50.
Mitochondrial CK (CK-Mi) is bound to the exterior surface of the inner mitochondrial membranes of muscle, brain, and liver. It migrates to a point cathodal to CK-MM and exists as a dimeric molecule of two identical subunits. It occurs in serum in both the dimeric state and in the form of oligomeric aggregates of high molecular weight (350,000). CK-Mi is not present in normal serum and is typically not present following MI. The incidence of CK-Mi ranges from 0.8% to 1.7%. For it to be detected in serum, extensive tissue damage must occur, causing breakdown of the mitochondrion and cell wall. Its presence does not correlate with any specific disease state but appears to be an indicator of severe illness. CK-Mi has been detected in cases of malignant tumor and cardiac abnormalities.
In view of the indefinite correlation between these atypical CK forms and a specific disease state, it appears that their significance relates primarily to the methods used for detecting CK-MB. In certain analytic procedures, these atypical forms may be measured as CK-MB, resulting in erroneously high CK-MB levels.
Methods used for measurement of CK isoenzymes include electrophoresis, ion-exchange chromatography, and several immunoassays, including radioimmunoassay (RIA) and immunoinhibition methods. Although mass methods are more sensitive and preferred for quantitation of CK-MB, electrophoresis has been the reference method. The electrophoretic properties of the CK isoenzymes are shown in Figure 13.5. Generally, the technique consists of performing electrophoresis on the sample, measuring the reaction using an overlay technique and then visualizing the bands under ultraviolet light. With electrophoresis, the atypical bands can be separated, allowing their detection apart from the three major bands. Often a strongly fluorescent band appears, which migrates in close proximity to the CK-BB form. The exact nature of this fluorescence is unknown, but it has been attributed to the binding of fluorescent drugs or bilirubin by albumin.
In addition to visualizing atypical CK bands, other advantages of electrophoresis methods include detecting an unsatisfactory separation and allowing visualization of adenylate kinase (AK). AK is an enzyme released from erythrocytes in hemolyzed samples and appearing as a band cathodal to CK-MM. AK may interfere with chemical or immunoinhibition methods, causing a falsely elevated CK or CK-MB value.
Ion-exchange chromatography has the potential for being more sensitive and precise than electrophoretic procedures performed with good technique. On an unsatisfactory column, however, CK-MM may merge into CK-MB and CK-BB may be eluted with CK-MB. Also, macro-CK may elute with CK-MB.
Antibodies against both the M and B subunits have been used to determine CK-MB activity. Anti-M inhibits all M activity but not B activity. CK activity is measured before and after inhibition. Activity remaining after M inhibition is a result of the B subunit of both MB and BB activity. The residual activity after inhibition is multiplied by 2 to account for MB activity (50% inhibited). The major disadvantage of this method is that it detects BB activity, which, although not normally detectable, will cause falsely elevated MB results when BB is present. In addition, the atypical forms of CK-Mi and macro-CK are not inhibited by anti-M antibodies and also may cause erroneous results for MB activity.
Immunoassays detect CK-MB reliably with minimal cross-reactivity. Immunoassays measure the concentration of enzyme protein rather than enzymatic activity and can, therefore, detect enzymatically inactive CK-MB. This leads to the possibility of permitting detection of infarction earlier than other methods. A double-antibody immunoinhibition assay is also available. This technique allows differentiation of MB activity due to AK and the atypical isoenzymes, resulting in a more specific analytic procedure for CK-MB.17 Point-of-care assay systems for CM-MB are available but not as widely used as those for troponins.
Assay Enzyme Activity
As indicated by Equation 13-4, CK catalyzes both forward and reverse reactions involving phosphorylation of creatine or ADP. Typically, for analysis of CK activity, this reaction is coupled with other enzyme systems, and a change in absorbance at 340 nm is determined. The forward reaction is coupled with the pyruvate kinase–LDH–NADH system and proceeds according to Equation 13-5:
 (Eq. 13-5)
(Eq. 13-5) The reverse reaction is coupled with the hexokinase–glucose-6-phosphate dehydrogenase–NADP system, as indicated in Equation 13-6:
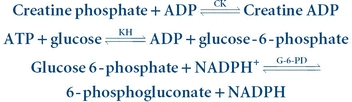 (Eq. 13-6)
(Eq. 13-6) Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


