Fig. 10.1
Jablonski diagram to illustrate the electronic changes occurring in the donor and acceptor molecules during a BRET process. The donor (D) molecule is normally in its ground state energy level. Upon substrate oxidation, it is elevated to a higher energy state (D*). Then, the excited donor loses some of its energy as vibrational relaxation energy. A part of the remaining energy is either emitted out as donor luminescence or is transferred to the nearby acceptor in a non-radiative manner. The acceptor fluorophore now gets excited (A*) and then relaxes back to its ground state by releasing photons at its characteristic wavelength, completing a BRET process
10.2.4 Factors Controlling Energy Transfer in RET
Several variables affect the efficiency of energy transfer between the two chromophore groups. First, the distance between the donor and acceptor molecule which is inversely proportional to the extent of energy transfer; the energy transfer gradually decreases when the distance between the donor and acceptor increases. The optimal distance for Förster resonance energy transfer is 1–10 nm. This is because the efficiency of energy transfer (E RET) is inversely proportional to the sixth power of the distance (R) between donor and acceptor molecules, Eq. 10.1.
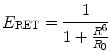
where R0 is the distance for 50 % energy transfer from the donor to the acceptor, which is about 5 nm [10, 21, 22]. Now, the Förster distance of a pair depends on the overlap integral of the donor emission spectrum with the acceptor absorption spectrum and their mutual molecular orientation and is calculated based on the Eq. 10.2.
![$$ \mathop R\nolimits_{0} = 2.11 \times 10^{ - 2} \times \left[ {\kappa^{2} \times J\left( \lambda \right) \times \eta^{ - 4} \times Q_{D} } \right]^{1/6} $$](/wp-content/uploads/2017/03/A305812_1_En_10_Chapter_Equ2.gif)
where 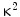 is the dipole orientation factor, η is the refractive index of the medium, J(λ) is the spectral overlap integral, and Q D denotes the donor quantum yield in the absence of acceptor. Because of the sixth-root dependence in the calculation of R 0, small changes in the value of Q D should not have a large effect on the overall BRET efficiency [23, 24]. Second, the orientation angle between donor and acceptor molecules affects RET efficiency since energy transfer will take place only if the transient dipoles of the interacting molecules are aligned in a position suitable for this transfer. Third, the degree of spectral overlap between donor emission and acceptor absorbance is also a significant factor. Higher the spectral overlap, the better the energy transfer [22]. Fourth, the quantum yield of the donor should be high, since the energy cannot be transferred if it is lost too quickly through non-radiative decay [6].
is the dipole orientation factor, η is the refractive index of the medium, J(λ) is the spectral overlap integral, and Q D denotes the donor quantum yield in the absence of acceptor. Because of the sixth-root dependence in the calculation of R 0, small changes in the value of Q D should not have a large effect on the overall BRET efficiency [23, 24]. Second, the orientation angle between donor and acceptor molecules affects RET efficiency since energy transfer will take place only if the transient dipoles of the interacting molecules are aligned in a position suitable for this transfer. Third, the degree of spectral overlap between donor emission and acceptor absorbance is also a significant factor. Higher the spectral overlap, the better the energy transfer [22]. Fourth, the quantum yield of the donor should be high, since the energy cannot be transferred if it is lost too quickly through non-radiative decay [6].
(10.1)
(10.2)
is the dipole orientation factor, η is the refractive index of the medium, J(λ) is the spectral overlap integral, and Q D denotes the donor quantum yield in the absence of acceptor. Because of the sixth-root dependence in the calculation of R 0, small changes in the value of Q D should not have a large effect on the overall BRET efficiency [23, 24]. Second, the orientation angle between donor and acceptor molecules affects RET efficiency since energy transfer will take place only if the transient dipoles of the interacting molecules are aligned in a position suitable for this transfer. Third, the degree of spectral overlap between donor emission and acceptor absorbance is also a significant factor. Higher the spectral overlap, the better the energy transfer [22]. Fourth, the quantum yield of the donor should be high, since the energy cannot be transferred if it is lost too quickly through non-radiative decay [6].In the RET-based PPI assay, i.e., BRET or FRET, the two chromophores are genetically tagged to two proteins whose interaction is to be investigated. The strict dependence of RET on the inter-chromophoric distance (1–10 nm) makes it an appropriate “molecular yardstick” for determining PPIs. This is true, since the average protein radius is ~5 nm, which means that a positive RET signal will only take place if the two proteins come within ~10 nm of each other, a distance that is an indicator of direct interaction between the two proteins [21].
10.2.5 Bioluminescence Resonance Energy Transfer
BRET exploits non-radiative (dipole–dipole) energy transfer occurring between a luminescent luciferase donor (instead of a fluorescent donor as in FRET) and a compatible fluorescent protein (FP) acceptor in order to study PPIs between two proteins fused to these donor and acceptor moieties (Fig. 10.2). The detection of a positive BRET signal means that the two proteins are situated within the BRET-permissive distance of 10 nm, thereby positively affirming their interaction [25, 26]. However, absence of a BRET signal does not necessarily mean that the two target proteins do not interact with each other. Lack of a signal can be accounted for by an unfavorable orientation between the donor and acceptor dipoles [27]. To nullify this issue, both N- and C-terminus fusions using flexible peptide linkers (6–18 amino acids) must be tested. Such design can reduce steric hindrance and improve the orientation. Normally, the linker sequence comprises of GGGGS or (GGGGS) × 2 residues where glycine–serine combinations confer flexibility, allowing the fusion proteins to fold to their optimal conformation [28, 29] and achieve the proximity distance between donor and acceptor.
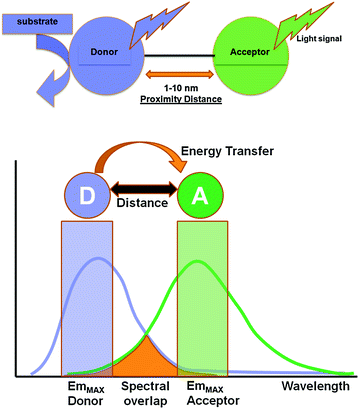
Fig. 10.2
Schematic illustration of the basic elements involved in a typical BRET system. In the presence of its specific substrate, the donor luciferase gets excited and emits light at its characteristic wavelength. Within the proximity distance of 1–10 nm, resonance energy transfer occurs to a suitable fluorescence acceptor, leading to the photon emission at its characteristic wavelength. Light signal obtained from both donor and acceptor emission can be measured using suitable band-pass filters, and BRET ratio can be judged as a measure of the distance between the donor and acceptor pair
BRET is an intrinsic phenomena occurring in the organisms Renilla reniformis and Aequorea victoria. In Aequorea victoria, a Ca2+-dependent photoprotein called aequorin releases flashes of blue light, which is transduced by a green fluorescent protein (GFP) to emit green light. Exploiting the underlying principle of BRET from Aequorea victoria, Xu et al. developed a BRET system in 1999 to study the interactions of circadian clock proteins in bacteria [25]. This BRET system termed as BRET1 uses Renilla luciferase (RLuc) as the donor moiety and enhanced yellow fluorescent protein (EYFP) as the acceptor (Emmax ~ 530 nm) moiety. The spectral resolution (separation of peak donor and acceptor emission spectra) achieved in BRET1 is ~50 nm only (Fig. 10.3) [23, 30].
Fig. 10.3
Schematic representations of BRET1 and BRET2 assays showing all the essential components in each. While both BRET1 and BRET2 use RLuc donor, they use different coelenterazine analogs such as Clz and Clz400 as indicated. Note the change in donor emission maximum in case of BRET2. In general, an 18-amino-acid linker is placed between the donor and acceptor proteins. Spectral resolution of each system is marked as a bidirectional arrow and spectral overlap region is marked with purple-color zone at the intersection of the donor and acceptor emission. Note, the donor and acceptor emission spans and intensities are thematic (not normalized)
Another BRET system, designated as BRET2 was developed that combined RLuc with a UV-excitable GFP variant called GFP2 [31, 32]. In this case, the substrate for RLuc was a coelenterazine analog DeepBlueC™ (also known as coelenterazine 400a/Clz400, bisdeoxycoelenterazine), which is similar to the native substrate in being cell permeable and non-toxic but shifts the Emmax to 400 nm. GFP2 excites at a maximum (Exmax) of 396 nm and emits photons at 510 nm. This yields a much larger spectral resolution of 110 nm (Fig. 10.3), which allows the use of wideband filters and minimizes bleed-through signal (residual emission of RLuc detected by the acceptor filter or vice versa). On the other hand, possible disadvantages associated with the use of the Clz400 substrate are poor substrate utilization, reduced quantum yield (~100 fold lower than Clz), and rapid decay in serum [23, 27]. Seeing the above two examples, it becomes clear that the balance between quantum yield and spectral resolution is of critical importance while choosing an appropriate BRET pair for in vivo applications. One should bear in mind that the same factors that affect the efficiency of RET are also applicable in BRET. To design the required BRET fusion proteins, the cDNA of the target proteins is inserted in frame into a suitable expression vector (by placing an appropriate linker as mentioned before) that has either the donor or acceptor gene. The stop codon of the amino terminus protein of the fusion partner should be removed by mutagenesis, and it should only be present in frame at the end of the carboxyl terminal protein so that the fusion protein can be expressed. One can design both C- and N- terminal protein fusions to assess their efficacy. Once the required fusion proteins are verified by expressing in mammalian cells, the BRET signal can be measured using suitable filter sets as explained later in this chapter. The BRET ratio [33] can be calculated as per Eqs. 10.3 and 10.4.
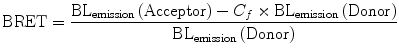
where

(10.3)
(10.4)
In Eq. 10.4, the correction factor (C f ) represents the BRET signal detected from cells transfected only with the donor protein. Subtracting this factor from the overall BRET ratio can give us an idea of the dynamic range for a particular BRET pair. Moreover, since BRET-based assays can be performed on live cells, under normal culture conditions and the indicative result being ratiometric, any variability due to assay volume or cell number variation is nullified.
10.3 Engineering the BRET Components
So far, we have just begun to understand the basic principles underlying the BRET technique. In order to explore the engineering aspects of BRET, we need to delve deeper into four main facets viz., the donor components i.e., the luciferase protein and its substrate, the acceptor fluorescent protein as well as detection devices (discussed in Sect. 10.5).
10.3.1 Bioluminescence Donor
As indicated before, to date, the luciferase enzyme from Renilla sp. has been predominantly used as the donor protein for the BRET systems in use, but in the recent years, several other luciferases from various other sources have also been engineered and used as BRET donors. We will now discuss various aspects of protein engineering that have been applied to each of them. Simultaneously, where applies, their respective engineered substrate is also discussed.
10.3.1.1 Renilla Luciferase and Coelenterazine
The main strength of a BRET signal is primarily dependent upon the efficiency of the luciferase enzyme to yield a high photon output upon the oxidation of its substrate. The 35-kDa RLuc protein typically emits light in the blue-green region (Emmax 480 nm). To further increase the donor quantum output, Angers et al. used coelenterazine-h (Clz-h) as a substrate for RLuc, which has the same Emmax as native Clz, but has a higher luminescence capacity than the latter [34]. For usage of this luciferase enzyme in mammalian systems, predominantly human cell lines, several commercial sources have developed a codon-optimized version hRLuc (BioSignal Packard) or hRL (Promega) that display improved bioluminescence than the native protein. However, a major limitation of hRLuc is its poor stability in serum (half-life of ~0.5–1 h in murine serum) which impedes bioluminescence or BRET imaging in animals for a sustained time period. To correct this problem, Loening et al. adopted a consensus-guided mutagenesis approach (for details, refer to Chap. 8) to identify beneficial mutations in hRLuc that render increased stability to the enzyme [35]. In the process of doing so, they also pinpointed mutations that showed increased quantum output. Upon introducing various point mutations, RLuc2 (C124A, M185V) and an eight mutation form of RLuc i.e., RLuc8 (A55T, C124A, S130A, K136R, A143M, M185V, M253L, and S287L) were developed that displayed greatly improved characteristics suitable for developing new BRET partners. Compared with the native enzyme, RLuc8 exhibited an intracellular half-life >50 h, a fourfold increment in quantum output, and a 5-nm wavelength shift in the emission spectrum [35]. The successful utilization of these mutations in improving BRET2 assay sensitivity with expanded dynamic range was demonstrated by us [24]. Later, by combining RLuc8 with the monomeric red FP variant mOrange (Emmax 564 nm), the BRET3 system was also developed (Fig. 10.4). On addition of Clz substrate, BRET3 provides a spectral resolution of ~85 nm [30]. Further, RLuc8 has shown to transfer adequate energy over a spectral distance of 100 nm when we combine RLuc8 donor with TagRFP (Emmax 584 nm) [36].
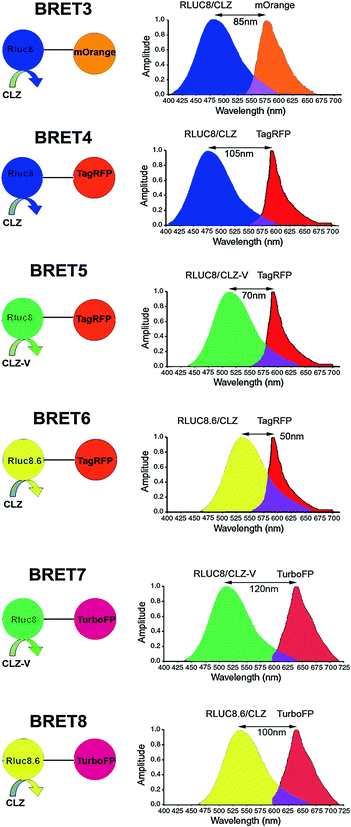
Fig. 10.4
Schematic representation of the expanded BRET fusion constructs developed using Renilla luciferase donor. The bioluminescence spectra illustrate the emission spectra of the RLuc mutants used as donor and the emission of the red fluorescent acceptor proteins. For all constructs, an 18-amino-acid linker is used between the donor and acceptor proteins. Luciferase substrates used in each case are indicated as either CLZ or CLZ-v. Spectral resolution of each system is marked as a bidirectional arrow. Spectral overlap region is also marked with a purple-color zone at the intersection of the emission spectrum. Note, the donor and acceptor emission spans and intensities are thematic (not normalized). Figure adapted with modifications as permitted by PNAS [36]
Another obstacle to the use of RLuc and its variants in BRET imaging was the blue-green light emission that greatly attenuated the signal in animal tissues, as pigments like myoglobin and hemoglobin strongly absorb photons from the blue-green region. Thus, use of RLuc for BRET detection from deeper tissues was severely limited [30, 37]. In the field of in vivo imaging, it is well known that light emitted above 600 nm can be better detected from deeper tissues of small animals. To achieve red-shifted light emission from RLuc, efforts have been made in two possible ways, i.e., genetic engineering of luciferase protein itself and chemical engineering of Clz substrate. Applying the genetic engineering approach to produce more red-shifted RLuc, Loening et al. [38] identified several mutants of which RLuc8.6 with an Emmax 535 nm (A123S, D154M, E155G, D162E, I163L, V185L point mutations in RLuc8) showed significant promise in live cells as well as animals. In 2010, Loening et al. [39] created another variant i.e., RLuc7 (Emmax 521 nm) with a rapid turnover and a half-life (~6.8 h) similar to the native RLuc in order to detect transient changes in gene expression. RLuc7 exhibited a twofold increase in the signal output compared to RLuc. Depending on the experimental requirements, one can decrease the stability of the luciferase enzyme without any mutagenesis by simply appending a signaling sequence at the 5′ or 3′ termini of the protein to direct the protein to early degradation. One such sequence is the PEST (Pro-Glu-Ser-Thr) sequence that has been frequently used to produce destabilized luciferases [40, 41].
Another remarkable finding is that the utilization of coelenterazine analog Clz-v with any of the RLuc variants has an additive effect as it further shifts the Emmax to the right by 35 nm. As shown, by using Clz-v in combination with RLuc8.6 protein, one can obtain an Emmax at 570 nm [36]. However, this substrate is not commercially available because of its problematic purification process possibly owing to sensitivity under chromatographic conditions, and it exhibits an order of magnitude increase in background auto-chemiluminescence [39]. Stepanyuk et al. ligated Clz-v to the coelenterazine binding protein (CBP) from the organism Renilla muelleri which acts as a ‘protector’ for Clz-v that results in a marked improvement in the stability of this conjugated Clz-v at 37 °C in addition to a higher bioluminescence signal output (twofold) in comparison with the native Clz-v when used with a R. muelleri luciferase (RLuc-m) mutant [42]. Taking advantage of these mutated RLuc donor and Clz analogs, several new combinations of BRET using red and far-red FPs have also been made as summarized in Fig. 10.4, expanding the scope of multiplexed BRET imaging. Of these new assays developed, BRET8 displaying a spectral resolution of 100 nm was used to image live cells and animals in the far-red region of the visible spectra [36]. Point to be mentioned, in Fig. 10.4, we have renamed some of the recently developed BRET vectors, such as BRET4.1, BRET5, and BRET6 as BRET5, BRET6, and BRET8, respectively, while eliminating some others reported such as BRET3.1 or BRET6.1 as their low spectral resolution may impede spectral separation for BRET imaging application.
Efforts continued further on synthesis of new coelenterazine analogs with better bathochromic emission shifts. Recently, Giuliani et al. [43] developed a new series of red-shifted coelenterazine analogs and a novel approach to alter the photochemical properties of the light-emitter intermediates. By insertion of a C-8-bonded S atom, the substrate molecules favor the emergence of lower energy emitters in coelenteramide. This bathochromic effect was evident in the presence of either the phenyl or the phenol ring in C-6, indicating that the red-shift can occur regardless of whether the main emitter is the neutral or the amide–anion form of the molecule.
One problem associated with coelenterazine and its derivatives was its quick decay upon auto-oxidation in the presence of aqueous media of cultured cells. For instance, Clz reduces in concentration by 50 % within 17 min of its addition to cell media. This decay not only reduces the availability of Clz molecules in the media, but also increases the background signal, thereby decreasing the assay sensitivity. Moreover, this limits its use in BRET assays to cell lysates only and fails to empower BRET applications to catch the live cell dynamics. This is equally true for other Clz analogs including Clz400. To solve this problem, Levi et al. [44] reported a clever approach to protect the putative oxygenation sites of Clz400 substrate and demonstrated that depending on the protection modifications, long-term BRET2 monitoring is achievable. Similarly, another commercial source has developed two substrates EnduRen™ and ViviRen™ that can be used specifically for live cell imaging [45]. These are protected forms of coelenterazine that have esters or oxymethyl ethers added at the site of substrate oxidation. The intracellular esterases and lipases yield the active Clz-h upon hydrolytic cleavage of the protected forms. The absence of active Clz-h in the media significantly reduces the background signal due to auto-oxidation, and the half-life of Clz-h increases. EnduRen™ and ViviRen™ substrates have been designed to display different kinetics. EnduRen™ shows a slow but gradual increase in bioluminescence. It reaches its maximum photon output at 90 min and then emits a constant signal for over 24 h (Fig. 10.5). In contrast, ViviRen™ can generate a threefold brighter signal which is instantaneous but short-lived. Pfleger et al. [46] demonstrated the use of EnduRen™ in monitoring the agonist-induced interaction of GPCRs in real time over a period of several hours. This was the first successful step in the direction of real-time BRET assays, as previously, the short half-life of Clz-h hampered the monitoring of PPIs in live cells. This modified BRET system was designated as extended BRET (eBRET), which is a superior approach for dynamic monitoring of PPI kinetics in their native cellular environment.
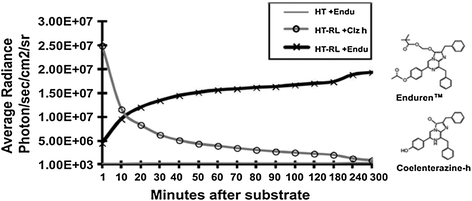
Fig. 10.5
Chart represents time kinetics of the light output after addition of normal coelenterazine-h substrate or protected coelenterazine-h substrate i.e., EnduRen™ [45]. Plain HT1080 mammalian cells (HT) or HT1080 cells overexpressing RLuc8 protein (HT-RL) were used for measuring the photon output as marked. Note the difference of time kinetics, where Clz-h substrate activity diminishes quickly within an hour; EnduRen™ substrate activity shows gradual increase till 90 min and remains maintained till the end point of measurement. Also note the y-axis scale bar representing a log scale. Note changes in the chemical structures that convert the Clz-h substrate into EnduRenTM
10.3.1.2 Firefly Luciferase and D-Luciferin
Another commonly used luciferase is the North American firefly luciferase (FLuc) from Photinus pyralis. It is a 61-kDa protein that emits orange light at an Emmax 562 nm [47, 48]. The emission of orange bioluminescence is a positive feature for bioluminescence imaging (BLI); however, its use in BRET is limited thus far, owing to its bulky size and low photon output. Moreover, the obligate dependence of FLuc on Mg2+ and ATP as its cofactors also diminishes its worth as a BRET donor. Nonetheless, FLuc has been employed in several BRET applications in combination with red- and far-red-shifted FPs such as DsRed [49] and mKate variant [50], respectively, as well as non-protein fluorophores [51]. For example, Arai et al. reported the use of the coral red fluorescent protein (DsRed) (Exmax 558 nm; Emmax 583 nm) as acceptor in combination with FLuc [49]. However, the use of DsRed is not preferred owing to its propensity to tetramerize, and the FLuc-DsRed system will have a spectral resolution of <20 nm which is highly unfavorable due to substantial overlap between FLuc and DsRed emission peak, thus necessitating the use of signal correction factor. Recently, BRET comprising a thermostable mutant form of firefly luciferase from Photinus pyralis (Ppy WT-TS; Emmax 565 nm) along with a variant of mKate (S158A) (Emmax 610 nm) was developed by Branchini et al. [52]. In the literature, Cy3 and Cy3.5 fluorophores have been attempted to be combined with FLuc as BRET partners [51]. However, the main drawback of using a non-protein fluorophore is that it has to be chemically linked with the donor and must be efficiently delivered within the cell for signal detection. Taking all these factors into account, the use of FLuc in BRET systems still needs to be optimized. Similar to RLuc, the codon-optimized version of FLuc (hFLuc) for use in mammalian cells has been created by commercial sources. Caysa et al. [37] have developed a red-shifted mutant of hFLuc (Emmax 615 nm) by introducing a single point mutation S284T. Even though the light output of the mutant FLuc drops to 25 % of the native FLuc, the signal output detected from whole animals is considerably higher (threefold) than its native counterpart, possibly due to the reduced tissue attenuation attributed to the red-shift in emission to above 600 nm. Branchini et al. [53] have also developed several FLuc mutants Ppy GR–TS and Ppy RE–TS that possess an Emmax of 546 and 610 nm, respectively, and Ppy RE8 and Ppy RE9 that show an Emmax of 617 nm, with Ppy RE9 being the superior luciferase. However, to the best of our knowledge, these mutant forms have so far not been recruited in any BRET system. In the D-luciferin molecule, N- and S-heteroatoms exist in a particular electron-rich configuration, which may play a fundamental role in the formation of multiple oxyluciferin excited states, resulting in a relatively broad emission spectrum [54]. Furthermore, the relatively slower and stable emission kinetics makes it naturally suitable for kinetic measurements from live environment without chemical modifications to its structure.
10.3.1.3 Other Luciferases
A range of other luciferases are now available from organisms like Metridia longa, Metridia pacifica, Cypridina noctiluca, Oplophorus gracilirostris, click beetle, railroad tapeworm (Phrixothrix hirtus), Vargula hilgendorfii, Gaussia princeps, etc., most of which are marine organisms. Some of them are popularly used as reporter genes for BLI, but have limited applicability in BRET due to their undesirable characteristics. For instance, Metridia luciferases (MLuc) are secretory in nature [55, 56], while Oplophorus luciferase (OLuc) is a 106-kDa secretory protein composed of a dimeric repeat [57] which is too bulky for use in BRET. Some luciferases show promising features and have successfully been exploited in the development of functional BRET systems. These comprise of click beetle green luciferase (CBG) [58], Gaussia luciferase (GLuc) [59], and Vargula luciferase (VLuc) [60].
Gammon et al. [58] reported a new combination of click beetle green (CBG) luciferase as donor and tdTomato as acceptor. Gaussia luciferase (GLuc) is a 19.9-kDa secretory protein with an Emmax 480 nm that extends till 600 nm. A codon-optimized version of GLuc (hGLuc) offers several advantages over other luciferases. In addition to its small size, hGLuc is highly heat stable, strongly resistant to acidic and basic conditions, and generates a ~100-fold higher intracellular signal compared to FLuc and hRLuc. Furthermore, unlike hRLuc or FLuc, it can be used either as a secretory or as an intracellular reporter depending on the experimental needs [61]. These features can contribute to the robustness of GLuc-based BRET systems. Li et al. reported the development of GLuc-EGFP BRET assay for protease activity sensing in in vitro [59]. Like GLuc, Vargula luciferase (VLuc) is a secretory protein, but of higher size (62 kDa) [62] with an Emmax 460 nm. The VLuc-EGFP combination has been reported by Otsuji et al. [60]. Irrespective of its bulky size, this system offers similar benefits in terms of applicability as in GLuc.
NanoLuc™ (NLuc) is a mutant form obtained from the deep-sea shrimp Oplophorus luciferase (OLuc). OLuc is a heteromeric structure composed of two 19-kDa catalytic subunits (OLuc-19) and two 35-kDa subunits (OLuc-35) that provide stability to the 19-kDa subunits. When Inouye et al. attempted to use the isolated OLuc-19 as a luciferase, they found that it was highly unstable and poorly expressed in the absence of its 35-kDa counterpart [57]. Hall et al. envisaged the creation of a variant of this OLuc-19 that ultimately led to the generation of NLuc. To achieve this, OLuc-19 was subjected to site-directed and random mutagenesis in order to obtain a stable variant. Further, through a single round of random mutagenesis, they generated a variant C1A4E with eight mutations (A4E, Q11R, A33K, V44I, A54F, P115E, Q124K, and Y138I). In the next phase, several coelenterazine derivatives were screened in order to create a superior substrate for optimal bioluminescence. Twenty-four coelenterazine analogs were developed and then screened against a library of C1A4E variants. Of these, the C1A4E variant (Q18L, F54I, F68Y, L72Q, M75K, and I90V) showed a ~10-fold higher stability over C1A4E at 37 °C with the coelenterazine derivative 2-furanylmethyl-deoxy-coelenterazine (Furimazine). In the final phase of optimization, they used furimazine to screen for other beneficial mutations in OLuc-19 variants. These mutations (L27V, K33N, K43R, and Y68D) were then combined with the previous mutations to produce NanoLuc™. In total, 16 amino acid substitutions of the wild-type OLuc-19 resulted in NLuc, having Emmax 460 nm and an intracellular half-life of >15 h at 37 °C. Using furimazine as its substrate exhibits a >150-fold higher light output than both RLuc and FLuc, which makes it a very attractive candidate for BRET multiplexing. Despite the emission maxima of NLuc in the blue-green region, its sufficiently high quantum output with furimazine can be used to couple it with any of the available BRET acceptors currently used with RLuc8.
10.3.2 Fluorescence Acceptor
The other component of a BRET system is a fluorescent protein that can be compatibly partnered with the luciferase in use. A good FP entails several characteristics. First, it should express efficiently in a system without invoking any cellular toxicity. Second, it should produce a high signal-to-noise ratio. Third, the FP should have sufficient photostability so as to be imaged for the duration of the experiment. Fourth, if the FP is to be fused to another protein of interest, then it should not have a tendency to oligomerize. Finally, it should be resistant to environmental conditions like pH and temperature [63].
Having realized the versatility of FPs in a variety of biological applications such as imaging and FRET, wild-type GFP from the organism Aequorea victoria (also known as wtGFP or avGFP) isolated by Shimomura et al. was cloned by Prasher et al. in 1992 [64]. Thus began the era of “GFP-technology”. The chromophore of wtGFP consists of Ser/Thr65-Tyr66-Gly67, which is protected by a shell consisting of 11 strands of β-barrels and one α-helix. The N- and C-termini of GFP are flexible, whereas the β-barrel shell is well structured and rigid, which protects the chromophore group from the external environment [65]. However, it does not fold efficiently at 37 °C since the optimal temperature for its maturation is 28 °C [66], and it forms weak dimers that may result in the formation of artifacts [65]. Thus, several GFP variants have been created, of which EGFP (Enhanced GFP from Clontech), AcGFP (Clontech), TurboGFP (Evrogen), and Emerald (Invitrogen) have overcome some of the limitations of GFP.
Yellow fluorescent proteins (YFPs) provide an edge over GFPs as their emission maxima lies in the 500–550 nm range. The original enhanced yellow fluorescent protein (EYFP) was derived from wtGFP and is now obsolete owing to its high sensitivity to chloride ions and slower maturation at 37 °C. Overcoming this problem, several successors of EYFP are now available such as citrine and its monomeric version mCitrine [67], Venus [68], and YPet [69]. Of these, YPet displays the brightest signal and so provides an advantage over its competitors [69].
As discussed above, a major shortcoming of using GFPs or YFPs used in early generation of BRET assays is their photon attenuation in mammalian tissues. To fulfill this demand of FPs emitting at red and far-red range, screening of various animal resources was initiated. A major breakthrough was the isolation of DsRed—a red fluorescent protein (RFP) from coral Discosoma striata. The excitation and emission maxima of DsRed are at 558 and 583 nm, respectively [70]. Moreover, it exhibits several positive features such as elevated extinction coefficient, high quantum yield, resistance to pH variations, and resistance to photobleaching. DsRed, however, tends to form tetramers [71], which often results in the poor localization as well as artificial oligomerization that may consequently impair the functions of proteins to which it is tagged. Furthermore, this chromophore matures very slowly, often taking days for the protein to convert from the premature greenish to the mature red emission. Monomeric RFP1 (mRFP1) [72] obtained after multiple rounds of mutagenesis (33 mutations in the parental DsRed) was the first true monomeric RFP with distinct improvements over previous versions of DsRed, namely DsRed2, T1 [73] and dimer2 [72], such as faster maturation and a 25-nm wavelength shift in excitation and emission. However, mRFP1 failed to acquire images with high spatial and temporal resolution due to its much lower quantum yield and extinction coefficient and a poor photostability [72]. Overcoming these limitations, Shaner et al. [74] and Wang et al. [75] reported a wide range of monomeric ‘fruit’ FPs named after fruits, representing the color similar to their emission such as mHoneydew, mBanana, mOrange, mTangerine, mStrawberry, mCherry [74], mRaspberry, and mPlum [75], as well as a tandem dimeric FP tdTomato, all with relatively long excitation and emission wavelengths (Table 10.1).
Table 10.1
The key properties of various classes of fluorescent proteins
Class | Fluorescent protein | Exmax (nm) | Emmax (nm) | Oligomerization | Reference |
|---|---|---|---|---|---|
Green | GFP | 395/475 | 508/503 | w.d. | [80] |
EGFP | 488 | 507 | w.d. | [80] | |
Emerald | 487 | 509 | w.d. | [80] | |
Yellow | EYFP | 514 | 527 | w.d. | [63] |
Venus | 515 | 528 | w.d. | [68] | |
mCitrine | 516 | 529 | m.m. | [67] | |
YPet | 517 | 530 | w.d. | [69] | |
Orange | mBanana | 540 | 553 | m.m | [74] |
mKO | 548 | 559 | m.m. | [81] | |
mOrange | 548 | 562 | m.m. | [74] | |
Red and Far-red | TurboRFP | 553 | 574 | d.m. | [76] |
tdTomato | 554 | 581 | t.d. | [74] | |
DsRed | 558 | 583 | t.m. | [70] | |
TagRFP | 555 | 584 | m.m. | [76] | |
mTangerine | 568 | 585 | m.m. | [74] | |
mStrawberry | 574 | 596 | m.m. | [74] | |
mCherry | 587 | 610 | m.m. | [74] | |
mRaspberry | 598 | 625 | m.m. | [75] | |
mKate2 | 588 | 633 | m.m. | [78] | |
mKate | 588 | 635 | m.m. | [77] | |
TurboFP635/Katushka | 588 | 635 | d.m. | [77] | |
mPlum | 590 | 649 | m.m. | [75] | |
TagRFP657 | 611 | 657 | m.m. | [79] |
Another group, Merzlyak et al. [76], developed RFP variants from a red eqFP578 protein isolated from the sea anemone Entacmea quadricolor. Dimeric eqFP578 again underwent several rounds of random mutagenesis that consequently produced an enhanced dimeric FP called TurboRFP with Emmax 574 nm. TurboRFP displayed superior qualities over DsRed2 such as greater pH stability, faster maturation at 37 °C, and higher extinction coefficient and quantum yield. Despite being a dimer, TurboRFP does not form aggregates in cell. It was further subjected to site-directed mutagenesis (R162E, Q166D, S180N, F198V, and F200Y) that contribute to the dimeric nature of TurboRFP as well as random mutagenesis to stabilize the monomeric form. The final product obtained was TagRFP which is a monomer and has a 10-nm shift in its Emmax. In addition, it is almost 2–3 folds brighter than mCherry, with a good maturation rate and a very high pH stability (pKa = 4), making it suitable for use in acidic organelles as well. Furthermore, Shcherbo et al. [77] succeeded in generating a dimeric RFP called Katushka or TurboFP635, with excitation and emission peaks at 588 nm and 635 nm, respectively. It is characterized by fast maturation rate, high pH stability (pKa = 5.5), low toxicity in mammalian cells, and superior brightness in comparison with its counterparts in the 650–800 nm optical window. However, since Katushka was dimeric in nature, Shcherbo et al. also targeted to develop monomeric Katushka, called mKate. They used monomeric TagRFP as the starting template and modified the residues surrounding its chromophores group to resemble that of Katushka. Thus, mKate has similar spectral characteristics as Katushka, albeit with slightly lowered quantum yield and pH stability. Shcherbo et al. [78] further optimized mKate and developed mKate2. Adding the mutation S165A to mKate, which converted its crystal structure from the trans to the cis form, resulted in an mKate variant with higher brightness and lower pH dependence. They also added two beneficial mutations V48A and K238R, developing the final product mKate2 with enhanced brightness and improved pH stability, photostability, and maturation rate. It is threefold brighter than mKate and 10-fold brighter than mPlum. Morozova et al. [79] have also recently succeeded in developing an even further red-shifted variant of mKate called TagRFP657 with Exmax and Emmax at 611 and 657 nm, respectively.
10.4 Engineering BRET Sensors for Functional Measurement
Unlike many other assays such as PCA, BRET system provides unique opportunity to readily adapt the newly evolved donor and acceptor proteins with preferred characteristics. This feature of BRET led to the rapid expansion and growth in identifying new BRET formats which eventually widened the BRET applicability. In this section, we will focus on engineering efforts that have expanded the scope of BRET applications. To do this, we can categorize reports under two major subheadings i.e., genetic BRET systems and synthetic BRET systems and discuss various applications that they have been tested for.
10.4.1 Genetic BRET Systems
As stated above, several genetically encoded BRET systems comprising of different combinations of luminescent and FP variants have been developed over the last decade. Due to attenuation of photons below 600 nm wavelengths in biological tissues, the BRET1 and BRET2 systems are suboptimal for small animal imaging use. To overcome their short falls, many variants of BRET systems utilizing Renilla luciferase mutants (RLuc8 or RLuc8.6) with mOrange (Emmax 564 nm), TagRFP (Emmax 584 nm), or TurboFP635 (Emmax 635 nm) FPs were rapidly developed by us as described in the previous section (Table 10.2). All these efforts were made with the goal of finding an optimal BRET assay that is most suitable for animal imaging applications. Together, these systems also give an edge in performing multiplexed BRET assays in live environment. Many of these systems can now serve as a unified format for in vitro measurements as well as physiologically relevant BRET experiments using live cells and mice models.
Table 10.2
Comprehensive chart featuring key characteristics of the BRET assays developed using Renilla luciferase
Assay | Donor | Acceptor | Substrate | Important featuresa |
|---|---|---|---|---|
BRET1 | RLuc 480 nm (Improved version using RLuc2/RLuc8) | YFP/EYFP 535 nm | Clz/EnduRen™ | 1 Efficient BRET |
• Spectral resolution 55 nm
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|