INTRODUCTION
An acute wound results from the sudden loss of anatomic structure in tissue following the transfer of kinetic, chemical, or thermal energy. Functionally, an acute wound should pass predictably through the phases of wound healing to result in complete and sustained repair. Acute wounds typically occur in recently uninjured and otherwise normal tissue. Acute wound healing is timely and reliable, completing the entire process within 6-12 weeks. Most surgical wounds are acute wounds.
Wound healing fails in a chronic wound. The process of tissue repair is prolonged and pathologic. The usual mechanism is dysregulation of one of the phases of normal acute wound healing. Most often, healing arrest occurs in an inflammatory phase. This prolonged inflammatory phase may be due to wound infection or another form of chronic irritation. Tissue and wound hypoxia is the other important mechanism for the development of a chronic wound. Failed epithelialization due to repeat trauma or desiccation may also result in a chronic partial thickness wound. Surgeons may sharply convert a chronic wound into an acute wound.
GENERAL CONSIDERATIONS
Surgeons often describe wound healing as primary or secondary. Primary healing occurs when tissue is cleanly incised and anatomically reapproximated. It is also referred to as healing by primary intention, and tissue repair usually proceeds without complication. Secondary healing occurs in wounds left open through the formation of granulation tissue and eventual coverage of the defect by migration of epithelial cells. Granulation tissue is composed of new capillaries, fibroblasts, and a provisional extracellular matrix that forms at the base of the early wound. This process is also referred to as healing by secondary intention. Most infected wounds and burns heal in this manner. Primary healing is simpler and requires less time and tissue synthesis than secondary healing. A wound healing primarily repairs a smaller volume than an open wound healing secondarily. The principles of primary and secondary healing are combined in delayed primary closure, when a wound is left open to heal under a carefully maintained, moist wound healing environment for approximately 5 days and is then closed as if primarily. Wounds treated with delayed primary closure are less likely to become infected than if closed immediately because bacterial balance is achieved and oxygen requirements are optimized through capillary formation in the granulation tissue.
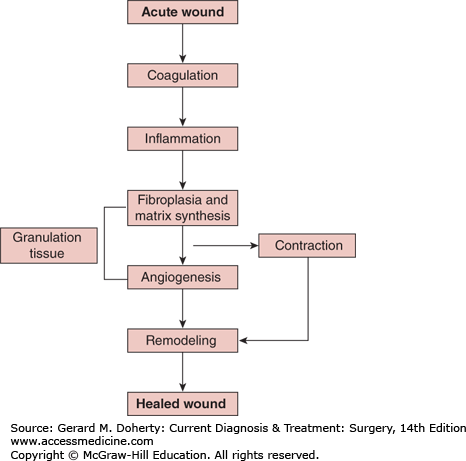
The complex process of wound healing normally proceeds from coagulation and inflammation through fibroplasia, matrix deposition, angiogenesis, epithelialization, collagen maturation, and finally wound contraction (Figure 6–1). Wound healing signals include peptide growth factors, complement, cytokine inflammatory mediators, and metabolic signals such as hypoxia and accumulated lactate. Many of these cellular signaling pathways are redundant and pleiotropic.
Figure 6–1.
Acute wound healing normally proceeds from coagulation and inflammation, through angiogenesis, fibroplasia, matrix deposition (granulation tissue formation), collagen maturation, epithelialization, and finally wound contraction. A chronic wound fails to heal anywhere along this wound healing pathway.
Following injury, a wound must stop bleeding in order to heal and for the injured host to survive. It is therefore not surprising that cellular and molecular elements involved in hemostasis also signal tissue repair. Immediately after injury, the coagulation products fibrin, fibrinopeptides, thrombin split products, and complement components attract inflammatory cells into the wound. Platelets activated by thrombin release insulinlike growth factor 1 (IGF-1), transforming growth factor α (TGF-α), transforming growth factor β (TGF-β), and platelet-derived growth factor (PDGF), which attract leukocytes, particularly macrophages, and fibroblasts into the wound. Damaged endothelial cells respond to a signal cascade involving the complement products C5a, tumor necrosis factor α (TNF-α), interleukin-1 (IL-1), and interleukin-8 (IL-8), and express receptors for integrin molecules on the cell membranes of leukocytes. Circulating leukocytes then adhere to the endothelium and migrate into the wounded tissue. Interleukins and other inflammatory components, such as histamine, serotonin, and bradykinin, cause vessels first to constrict for hemostasis and later to dilate, becoming porous so that blood plasma and leukocytes can migrate into the injured area.
The very early wound inflammatory cells increase metabolic demand. Since the local microvasculature is damaged, a local energy sink results, and Pao2 falls while CO2 accumulates. Lactate in particular plays a critical role, since its source is mainly aerobic, and its level is tightly regulated by tissue oxygen levels. Oxidative stress is an important signal for tissue repair. These conditions trigger reparative processes and stimulate their propagation.
Macrophages assume a dominant role in the synthesis of wound healing molecules as coagulation-mediated tissue repair signals fall. Importantly, macrophages, stimulated by fibrin, continue to release large quantities of lactate. This process continues even as oxygen levels begin to rise, thereby maintaining the “environment of injury.” Lactate alone stimulates angiogenesis and collagen deposition through the sustained production of growth factors. Unless the wound becomes infected, the granulocyte population that dominated the first days diminishes. Macrophages now cover the injured surface. Fibroblasts begin to organize, mixed with buds of new blood vessels. It has been shown that circulating stem cells, such as bone marrow–derived mesenchymal stem cells, contribute fibroblasts to the healing wound, but the extent of this process is as yet unknown.
Throughout wound healing, fibroplasia (the replication of fibroblasts) is stimulated by multiple mechanisms, starting with PDGF, IGF-1, and TGF-β released by platelets and later by the continual release of numerous peptide growth factors from macrophages and even fibroblasts within the wound. Growth factors and cytokines shown to stimulate fibroplasia and wound healing include fibroblast growth factor (FGF), IGF-1, vascular endothelial growth factor (VEGF), IL-1, IL-2, IL-8, PDGF, TGF-α, TGF-β, and TNF-α. Dividing fibroblasts localize near the wound edge, an active tissue repair environment with tissue oxygen tensions of approximately 40 mm Hg in normally healing wounds. In cell culture, this Pao2 is optimum for fibroblast replication. Smooth muscle cells are also likely progenitors because fibroblasts seem to migrate from the adventitia and media of wound vessels. Lipocytes, pericytes, and other cell sources may exist for terminal differentiation into repair fibroblasts.
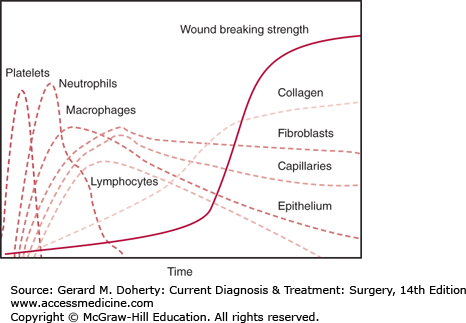
Fibroblasts secrete the collagen and proteoglycans of the connective tissue matrix that hold wound edges together and embed cells of the healing wound matrix. These extracellular molecules assume polymeric forms and become the physical basis of wound strength (Figure 6–2). Collagen synthesis is not a constitutive property of fibroblasts but must be signaled. The mechanisms that regulate the stimulation and synthesis of collagen are multifactorial and include both growth factors and metabolic inputs such as lactate. The collagen gene promoter has regulatory binding sites to stress corticoids, the TGF-β signaling pathway, and retinoids, which control collagen gene expression. Other growth factors regulate glycosaminoglycans, tissue inhibitors of metalloproteinase (TIMP), and fibronectin synthesis. The accumulation of lactate in the extracellular environment is shown to directly stimulate transcription of collagen genes as well as posttranslational processing of collagen peptides. It is clear that the redox state and energy stores of repair cells occupying the wound regulate collagen synthesis.
The increase in collagen messenger RNA (mRNA) leads to an increased procollagen peptide. This, however, is not sufficient to increase collagen deposition because procollagen peptide cannot be transported from the cell to the extracellular space until, in a posttranslational step, a proportion of its proline amino acids are hydroxylated. In this reaction, catalyzed by prolyl hydroxylase, an oxygen atom derived from dissolved O2 is inserted (as a hydroxyl group) into selected collagen prolines in the presence of the cofactors ascorbic acid, iron, and α-ketoglutarate. Thus, accumulation of lactate, or any other process that decreases the nicotinamide adenine dinucleotide (NAD+) pool, leads to production of collagen mRNAs, increased collagen peptide synthesis, and (provided enough ascorbate and oxygen is present) increased posttranslational modification and secretion of collagen monomers into the extracellular space.
Another enzyme, lysyl hydroxylase, hydroxylates many of the procollagen lysines. A lysyl-to-lysyl covalent link then occurs between collagen molecules, maximizing mature collagen fiber strength. This process, too, requires adequate amounts of ascorbate and oxygen. These oxygenase reactions (and therefore collagen deposition) are rate limited by tissue oxygen level, Pao2. The rates are half-maximal at about 20 mm Hg and maximal at about 200 mm Hg. Hydroxylation can be “forced” to supernormal rates by tissue hyperoxia. Collagen deposition, wound strength, and angiogenesis rates may be increased and accelerated as tissue Pao2 is elevated.
Angiogenesis is required for wound healing. It is clinically evident about 4 days following injury but begins earlier when new capillaries sprout from preexisting venules and grow toward the injury in response to chemoattractants released by platelets and macrophages. In primarily closed wounds, budding vessels soon meet and fuse with counterparts migrating from the other side of the wound, establishing blood flow across the wound. In wounds left open, newly forming capillaries connect with adjacent capillaries migrating in the same direction, and granulation tissue forms. Numerous growth factors and cytokines are observed to stimulate angiogenesis, but animal experiments indicate that the dominant angiogenic stimulants in wounds are derived first from platelets in response to coagulation and then from macrophages in response to hypoxia or high lactate, fibrin, and its products.
Epithelial cells respond to several of the same stimuli as fibroblasts and endothelial cells within the mesenchymal area of a wound. A variety of growth factors also regulate epithelial cell replication. TGF-α and keratinocyte growth factor (KGF), for instance, are potent epithelial cell mitogens. TGF-β tends to inhibit epithelial cells from differentiating and thus may potentiate and perpetuate mitogenesis, though it is itself not a mitogen for these cells. During wound healing, mitoses appear in the epithelium a few cells away from the wound edge. The new cells migrate over the cells at the edge and into the unhealed area and anchor to the first unepithelialized matrix position encountered. The Pao2 on the underside of the cell at the anchor point is usually low. Low Pao2 stimulates squamous epithelial cells to produce TGF-β, likely suppressing terminal differentiation and again supporting further mitosis. This process of epidermal-mesenchymal communication repeats itself until the wound is closed.
Squamous epithelialization and differentiation proceed maximally when surface wounds are kept moist. It is clear that even short periods of drying impair the process, and therefore wounds should not be allowed to desiccate. The exudates from acute, uninfected superficial wounds also contain growth factors and lactate, and therefore recapitulate the growth environment found at the base of the wound.
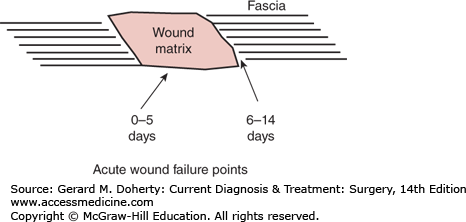
Remodeling of the wound extracellular matrix is also a well-regulated process. First, fibroblasts replace the provisional fibrin matrix with collagen monomers. Extracellular enzymes, some of which are Pao2-dependent, quickly polymerize these monomers, initially in a pattern that is more random than in uninjured tissue, predisposing early wound to mechanical failure. Progressively, the very early provisional matrix is replaced with a more mature one by forming larger, better organized, stronger, and more durable collagen fibers. The very early wound provisional matrix usually mechanically fails within the matrix itself (days 0-5). Next, mechanical failure occurs at the matrix-tissue interface or fusion point (Figure 6–3). The mechanism for connecting the wound matrix to the uninjured tissue border is poorly understood.
Reorganization of the new matrix is an important feature of healing, and fibroblasts and leukocytes secrete collagenases that ensure the lytic component. Turnover occurs rapidly at first and then more slowly. Even in simple wounds, wound matrix turnover can be detected chemically for as long as 18 months. Healing is successful when a net excess of matrix is deposited despite concomitant lysis. Lysis, in contrast to anabolic synthesis, is less dependent upon energy and nutrition. If synthesis is impaired, however, lysis weakens wounds.
During rapid turnover, wounds normally gain strength and durability but are vulnerable to contraction or stretching. Fibroblasts exert the force for contraction. Fibroblasts attach to collagen and each other and pull the collagen network together when the cell membranes shorten as the fibroblasts migrate. The wound myofibroblast, a specialized phenotype, expresses intracellular actin filaments that also contribute force to fibroblast-mediated wound contraction. The collagen fibers are then fixed in the packed positions by a variety of cross-linking mechanisms. Both open and closed wounds tend to contract if not subjected to a superior counterforce. The phenomenon is best seen in surface wounds, which may close 90% or more by contraction alone in loose skin. For example, the residual of a large open wound on the back of the neck may be only a small area of epithelialization. On the back, the buttock, or the neck, this is often a beneficial process, whereas in the face and around joints, the results may be disabling or disfiguring. Pathological wound contraction is usually termed a contracture or a stricture. Skin grafts, especially thick ones, may minimize or prevent disabling wound contractures. Dynamic splints, passive or active stretching, or insertion of flaps containing dermis and subdermis also counteract contraction. Prevention of a stricture often depends on ensuring that opposing tissue edges are well perfused so that healing can proceed quickly to completion and contraction stops. Healing wounds may also stretch during active turnover when tension overcomes contraction. This may account for the laxity of scars in ligaments of injured but unsplinted joints and the tendency for incisional hernia formation in abdominal wounds of obese patients.
Tissue other than skin heals generally by the same fundamental pathways. Although tissue structure may be specialized, the initial repair processes are shared. It does appear that the rate and efficiency of wound healing in different tissue types depends in large part on total collagen content, collagen organization, and blood supply.
The rate of repair varies from one part of the intestine to the other in proportion to blood supply. Anastomoses of the colon and esophagus heal least reliably and are most likely to leak, whereas failure of stomach or small intestine anastomoses is rare. Intestinal anastomoses regain strength rapidly when compared to skin wounds. After 1 week, bursting strength may exceed the uninjured surrounding intestine. However, the surrounding intestine also participates in the reaction to injury, initially losing collagen by lysis, and as a result may lose strength. For this reason, leakage can occur a few millimeters from the anastomosis. A tight suture line causing ischemia will exacerbate this surgical problem.
The mesothelial cell lining of the peritoneum also is important for healing in the abdomen and GI tract. The esophagus and retroperitoneal colon lack a serosal mesothelial lining, which may contribute to failed wound healing. There is evidence that mesothelial cells signal the repair of peritoneal linings and are a source of repair cells.
Comorbidities that delay collagen synthesis or stimulate collagen lysis are likely to increase the risk of perforation and leakage. The danger of leakage is greatest from the fourth to seventh days, when tensile strength normally would rise rapidly but may be impeded by impaired collagen deposition or increased lysis. Local infection, which most often occurs near esophageal and colonic anastomoses, promotes lysis and delays synthesis, thus increasing the likelihood of perforation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


