Fig. 11.1
Long-term blockade of cocaine-primed reinstatement responding. CocH vector-treated rats (N = 14) made significantly fewer responses (# p < 0.05) on the lever previously paired with cocaine than control rats (N = 26) following a 10 mg/kg cocaine i.p. priming injection during reinstatement testing sessions up to 24 weeks after vector or control injections. This effect was specific to cocaine, as 2 mg/kg d-amphetamine i.p. priming injections administered at 24 weeks post-vector injection elicited a similar number of responses in CocH vector-treated and control rats
The sustained and specific blockade of reinstatement was an encouraging sign that CocH gene transfer might eventually prove useful in protecting a treatment-seeking human from experiencing a major relapse into cocaine abuse triggered by a single re-encounter with that drug. A weakness in the simple reinstatement model, however, was that the “priming exposure” involved a relatively slow route of drug delivery (i.e., subcutaneous or intraperitoneal injection). Since the intensity of reward typically increases with the speed of drug delivery to the brain, more realistic experiments based on rapid drug delivery were clearly needed. The most rigorous test we could construct was to assess the effect of gene transfer upon ongoing responding for i.v. cocaine reward delivered through a cannula permanently implanted in the vena cava. Such experiments, carried out in Dr. Carroll’s laboratory, used two groups of rats fully trained on lever pressing for cocaine reward and maintaining a steady level of responding. These groups were given different doses of hdAD vector encoding CocH (1011 and 1012 viral genomes per rat). The results were clear cut (Fig. 11.2). With either dose there was a lag of several days during which ongoing responding continued at the same stable level as before treatment. Then at about 5 days, the lever-pressing behavior began to increase. This outcome occurred in both experimental groups. It was interpreted as an attempt to compensate for a loss of reward derived from the drug delivery, which was unchanged in the amount of cocaine. This compensatory behavior was apparently successful for the rats that received low-dose vector, which continued with their increased lever pressing indefinitely. In sharp contrast, the rats that received high-dose vector abandoned lever pressing altogether and for the entire remainder of the 2-month study.
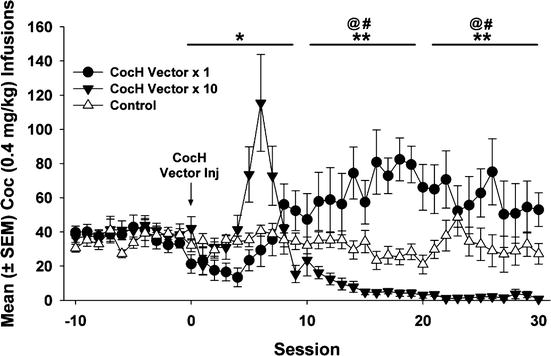
Fig. 11.2
Long-term blockade of ongoing responding for cocaine reward. Mean (± SEM) cocaine infusions before and after delivery of CocH vector or vehicle. CocH vector × 1 rats notably increased their infusions relative to controls (** p < 0.01) and baseline (at p < 0.01) following hdAD-CocH vector injection. While the CocH vector × 10 rats increased their infusions for 10 days following vector delivery, their infusions subsequently decreased to near-zero levels and were significantly lower than their infusions at baseline (at p < 0.01) and lower than that of CocH vector × 1 rats (** p < 0.01)
11.4 Avoiding Vector Toxicity
Despite these encouraging results, there are serious challenges to the clinical application of hdAD vectors. The greatest of these is the demonstrated risk of short term toxicity, particularly in the liver, induced by transient exposure to the capsid proteins in the viral vector. These proteins are required for initial interaction with hepatocytes, our preferred cell type for enzyme transduction, but they evoke a dose-related response from the innate host immune system, which can be rapid and severe (Brunetti-Pierri et al. 2004; Muruve 2004). Hepatocytes are particularly vulnerable during the process of vector uptake. This reaction is equivalent to the one provoked by early-generation E-1 deleted AD vectors, which is suspected as the primary cause of the fatal outcome in one of the earliest clinical trials of AD gene therapy (Raper et al. 2003). Since the immune response is transient and limited to the period during which viral capsid proteins remain in situ, the problem can be mitigated to an extent by temporary immune suppression. In fact, although multiple factors come into play, it has been reported that pretreatment with the anti-inflammatory steroid, dexamethasone (Seregin et al. 2009), reduces the innate toxicity of adenoviral coat proteins. We used this approach in studies with rhesus monkeys given relatively high doses of hdAD under thiofluorane anesthesia: up to 1012 viral particles per animal. None of the treated animals exhibited signs of discomfort or distress after recovering from the anesthesia or later on. Nonetheless, it would be premature to conclude that clinical application of such vectors is completely safe.
The principal alternative to hdAD in gene therapy for cocaine abuse is AAV. AAV vectors have seen limited applications because of the tight restrictions on packaging capacity, i.e., the length of DNA sequences that can be introduced as transgene payload is less than 5 kB as noted earlier. A second problem is that infections with wild type AAV are prevalent in the human population and the resulting immunity prevents effective use of vectors with the same serotype. On the positive side, AAV has never been associated with human illness, and it has proved relatively benign in animal studies. As a result, and despite the drawbacks just noted, several clinical trials have been completed with AAV vectors and more are ongoing or pending. In our early mouse experiments, however, AAV-8 vector loaded with cocaine hydrolase generated only 20 % as much plasma cocaine hydrolase activity as the equivalent hdAD vector. If this problem can be overcome, the stage will be set for a fair comparison between two promising vector platforms, in which the primary standard will be delivery of the greatest drug-metabolizing power with the risk of adverse effect.
11.4.1 Probable Duration of Expression in Humans
It should not be necessary to provide permanent gene transduction in order to treat addiction effectively. However, at least 6 months of effective drug interception is needed, and a period of 2 or 3 years may be desirable to aid in cessation of drug intake and avoidance of relapse. This time frame would not require a vector that incorporates into genomic DNA (with attendant risk of disrupting oncogenes), but can be achieved with a vector that persists as a quasi-stable episomal element (e.g., helper-dependent “gutted” adenoviral vector or adeno-associated viral vector). The latter two vectors show long persistence even though they do not propagate from cell to cell and are diluted or lost as a result of mitosis. Even in rats and mice, whose hepatocytes turn over with a half-life of a year or less, effective levels of transgene remain for many months after initial vector treatment. Life spans for hepatocytes in adult human liver are estimated to lie between 300 and 500 days (Spalding et al. 2005). Therefore, if cell turnover is actually the primary mechanism for loss of vector DNA, one could expect a therapeutic window on the order of a year or two at most.
11.4.2 Drawbacks and Obstacles to Gene Transfer
One major present drawback is a persistent concern for safety, which may be well founded and certainly deserves attention. A key risk factor appears to be the brief but intense innate immune response to viral capsid proteins (Muruve 2004). Much research has concentrated on this issue and how to minimize or avoid it (Brunetti-Pierri and Ng 2008, 2011). Deletion of multiple viral genes while preserving transduction capability is one important step, as in the engineered helper-dependent adenoviral vectors (Morral et al. 1999) or the vectors based on naturally helper-dependent adeno-associated virus (Brantly et al. 2009; Manno et al. 2006). These modified agents typically have no ability to express viral proteins in vivo and thus, longer term, may remain “hidden” from immune surveillance (Vetrini and Ng 2011). On the other hand, to deliver an engineered DNA payload, viral particles must first attach to the plasma membranes of host cells that can translate the genetic information into a protein product. This function requires packaging of the functional DNA within a coat that (a) protects it from dispersal and metabolic attack in body fluids, (b) selectively binds surface targets characteristic of the host cells chosen for transgene product, and (c) stimulates endocytosis of the delivered DNA. At the present state of vector technology, the most efficient means of accomplishing this objective uses artificial protein coats derived from the shell of a relatively benign “helper virus.” The immune system still recognizes these coats, which remain able to trigger a seriously adverse response. Such responses are typically short lived, however. They are also dose related and can be minimized by delivering a smaller load of vector, albeit at the price of obtaining less transgene product.
Fortunately, transient immunosuppression with anti-inflammatory steroids, such as dexamethasone, will reduce host immune responses to viral vector and lessen subsequent toxicity (Seregin et al. 2009) while also prolonging transgene expression (Kumahara et al. 2005). In some of our animal studies, immunosuppression was also required for adequate levels of therapeutic protein because the subjects had undergone prior procedures that established a state of chronic inflammation. This happened in animals subjected to procedures creating sustained inflammatory stress (e.g., implantation of multiple cannulas), which can impair subsequent virally mediated transgene expression (Gao, Brimijoin, 2015). In higher organisms, especially nonhuman primates and humans, the acute immune response to vector is very strong, associated with a sharp rise in cytokines such as interleukin 6, while immunosuppression is more difficult and less effective (Brunetti-Pierri et al. 2004). Progress in this arena is critical to future success.
11.5 Long-Term Safety Issues
In view of the high bar that must eventually be cleared if BChE/CocH gene transfer can be tested in clinical trial, our research group has carried out numerous studies to evaluate safety issues. These studies were carried out largely in mice but also to an extent in rats and in a few rhesus monkeys. These studies involved delivery of viral vector in doses up to 3 × 104 genomes per kg body weight in rodents and 3 × 1012 genomes per kg in rhesus (several times larger than would likely be given to a human patient). Multiple issues were examined, many of them selected in light of the fact that BChE gene transfer necessarily increases the capacity for hydrolysis of acetylcholine, the cholinergic neurotransmitter. For example, grip strength and maximum treadmill effort were evaluated in mice (Murthy et al. 2014a). Also, even though systemic gene transfer should not affect CNS function and our prior studies showed no viral particles in the brains of treated rodents, cognitive function was tested with mice in a structured water maze. No deficits were observed. Liver function appeared normal and there was no elevation of sentinel enzymes such as alanine aminotransferase in plasma. Resting mouse blood pressure was normal as measured by semiautomatic plethysmography. EKG telemetry with indwelling sensors revealed no alteration of resting heart rate or QT interval (unpublished data). Spontaneous locomotor activity was not affected, nor was body temperature, O2–CO2 exchanger, or overall metabolic rate as assessed from 24-h sessions in a metabolic chamber (Murthy et al. 2014a, b). And finally, no differences between treated and untreated mice were detected in a pathology analysis of multiple tissues including the liver, kidney, heart, lung, brain, muscle, stomach, and small intestine, in professional harvests at 1 week, 3 weeks, 3 months, and 22 months after vector delivery.
11.6 FDA Concerns
When it comes to protein-based therapeutics, the US FDA appears to have several general concerns focusing on duration of action, immunological responses, and amount of protein delivery over time, among other issues. A gene therapy approach necessarily complicates the process of seeking approval for an investigational therapy of this nature. There are two key reasons. First, the vector is seen as a potential risk in itself, regardless of the nature of encoded protein, and second, the magnitude and time course of the vector-transduced protein is more difficult to predict and control as compared with direct administration of a highly purified final product (enzyme). In fact, after a recent pre-IND conference with the FDA, the agency explicitly stated concerns about the duration, level, and enhanced affinity of our proposed transgene expression product (i.e., cocaine hydrolase) which could persist for years in subjects receiving this investigational treatment (viral vector). Based on those concerns, the FDA asked for extensive new data demonstrating vector biodistribution and persistence, as well as transgene expression levels over time. The agency also expressed a need for supporting data addressing the potential that over-expression of this enzyme and its resulting effect on endogenous biochemical pathways (e.g., cholinergic synapses) and clinical medications (e.g., pharmacological muscle relaxants, morphine-based pain medication, antibiotics, etc.) that might be administered to these subjects over the course of their lifetime. Proponents of a gene therapy for cocaine addiction must take all possible steps to address such issues. What the FDA did not pose as a precondition for eventual clinical trial was to duplicate all preclinical testing on vector transduction with essentially identical tests based upon purified CocH. In short, the focus is upon the candidate therapeutic (i.e., viral vector) and not the downstream enzyme product. Our attempts to allay all concerns expressed in regard to BChE gene transfer are extensive and ongoing. They are briefly summarized below in the interests of guiding other investigators with similar goals and issues.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


