Chapter 15 Viral, fungal, protozoal and helminthic infections
• Viruses present a more difficult problem of chemotherapy than do higher organisms, e.g. bacteria, for they are intracellular parasites that use the metabolism of host cells.1 Highly selective toxicity is, therefore, harder to achieve. In the past 15 years, identification of the molecular differences between viral and human metabolism has led to the development of many effective antiviral agents; four were available in 1990, now there are over 40.
• Fungal infections range from inconvenient skin conditions to life-threatening systemic diseases; the latter have become more frequent as opportunistic infections in patients immunocompromised by drugs or AIDS, or in those receiving intensive medical and surgical interventions in intensive care units.
• Protozoal infections. Malaria is the major transmissible parasitic disease in the world. Drug resistance is an increasing problem and differs with geographical location, and species of plasmodium.
• Helminthic infestations cause considerable morbidity. The drugs that are effective against these organisms are summarised.
Viral infections
Antiviral agents are most active when viruses are replicating. The earlier that treatment is given, therefore, the better the result. Apart from primary infection, viral illness is often the consequence of reactivation of latent virus in the body. Patients whose immune systems are compromised may suffer particularly severe illness. Viruses are capable of developing resistance to antimicrobial drugs, with similar implications for the individual patient, for the community and for drug development. An overview of drugs that have proved effective against virus diseases appears in Table 15.1.
Table 15.1 Drugs of choice for virus infections
| Organism | Drug of choice | Alternative |
|---|---|---|
| Varicella zoster | ||
| chickenpox | Aciclovir | Valaciclovir or famciclovir |
| zoster | Aciclovir or famciclovir | Valaciclovir |
| Herpes simplex | ||
| keratitis | Aciclovir (topical) | |
| labial | Aciclovir (topical and/or oral) | Valaciclovir or famciclovir |
| genital | Aciclovir (topical and/or oral) | Valaciclovir |
| Famciclovir (oral) | Penciclovir | |
| encephalitis | Aciclovir | |
| disseminated | Aciclovir | Foscarnet |
| Human immunodeficiency virus (HIV) | Lamivudine/emtricitabine | Abacavir |
| Tenofovir | Didanoside | |
| Zidovudine | Stavudine | |
| Lopinavir/ritonavir | Saquinavir | |
| Atazanavir | Darunavir | |
| Fosamprenavir | Tipranavir | |
| Efavirenz | Nevirapine | |
| Etravirine | ||
| Raltegravir | ||
| Enfuvirtide | ||
| Maraviroc | ||
| Hepatitis B | Pegylated interferon α-2a and interferon 2b, lamivudine | Adefovir, tenofovir, entecavir, telbivudine |
| Hepatitis C | Pegylated interferon α-2a or interferon 2b plus ribavirin | |
| Hepatitis D | Interferon-α | Pegylated interferon α-2a and interferon 2b |
| Influenza A | Zanamivir, oseltamivir | Amantadine |
| Cytomegalovirus (CMV) | Valganciclovir, ganciclovir | Foscarnet, cidofovir |
| Respiratory syncytial virus | Ribavirin | Palivizumab |
| Papillomavirus (genital warts) | Imiquimod | |
| Molluscum contagiosum | Imiquimod | Cidofovir |
Herpes simplex and varicella zoster
Aciclovir
• Skin infections, including initial and recurrent labial and genital herpes, most effective when new lesions are forming; skin and mucous membrane infections (as tablets or oral suspension).
• Ocular keratitis (topical treatment with ophthalmic ointment is standard, oral treatment is also effective).
• Prophylaxis and treatment in the immunocompromised (oral, as tablets or suspension).
Aciclovir-resistant herpes simplex virus has been reported in patients with AIDS but remains rare in immunocompetent patients. Foscarnet (p. 221) and cidofovir (p. 221) have been used in these cases.
• Chickenpox, particularly in the immunocompromised (i.v.) or in the immunocompetent with pneumonitis or hepatitis (i.v.).
• Shingles in immunocompetent persons (as tablets or suspension, and best started within 48 h of the appearance of the rash). Immunocompromised persons will often have more severe symptoms and require i.v. administration.
Human immunodeficiency virus (HIV)
General comments
• The aims of antiretroviral therapy are to delay disease progression and prolong survival by suppressing the replication of the virus. Optimal suppression also prevents the emergence of drug resistance and reduces the risks of onward transmission to sexual partners and the unborn children of HIV-infected mothers. Virological failure may be defined as primary where there is inability to reduce plasma HIV viral load to fewer than 50 copies per microlitre despite 6 months of antiretroviral therapy, or secondary if there is failure to maintain viral load suppression at less than 50 copies per microlitre.
• No current antiviral agents or combinations eliminate HIV infection, but the most effective combinations (so-called ‘highly active antiretroviral therapy’, HAART) produce profound suppression of viral replication in many patients and allow useful reconstitution of the immune system, measured by a fall in the plasma viral load and an increase in the numbers of cytotoxic T cells (CD4 count). Rates of opportunistic infections such as Pneumocystis carinii pneumonia and cytomegalovirus (CMV) retinitis are reduced when CD4 counts are restored, and life expectancy is markedly increased.
• Combination therapy reduces the risks of emergence of resistance to antiretroviral drugs, which is increasing in incidence even in patients newly diagnosed with HIV. Mutations in the viral genome either prevent binding of the drug to the active site of the protease or reverse transcriptase enzymes, or lead to removal of the drug from the reverse transcriptase active site. The potential for rapid development of resistance is immense because untreated HIV replicates rapidly (50% of circulating virus is replaced daily), the spontaneous mutation rate is high, the genome is small, the virus will develop single mutations at every codon every day, and for many antiretroviral agents a single mutation will render the virus fully resistant.
• The decision to begin antiretroviral therapy is based primarily on the CD4 cell count (most current recommendations are to start in patients with counts below 350 cells per microlitre). Early initiation of antiretroviral therapy should also be considered for patients with CD4 cell count above 350 cells per microlitre but a low CD4 percentage (e.g < 14%), those with an AIDS diagnosis (e.g. Kaposi sarcoma), hepatitis B and HIV co-infection where treatment is indicated, and in conditions where achieving a suppressed viral load is desired in order to prevent transmission (e.g. in pregnancy).
• There are currently more than 20 approved antiretroviral agents in four classes, plus various fixed drug combinations (Table 15.2).
• Current HAART regimens use a combination of drugs that act at different phases of the viral life cycle. The most frequently used combinations employ a backbone of two nucleoside analogue reverse transcriptase inhibitors (NRTIs) plus either a non-nucleoside reverse transcriptase inhibitor (NNRTI) or a ritonavir-boosted protease inhibitor (rPI). The choice for the individual patient is best made after reference to contemporary, expert advice (see the websites listed in the Guide to further reading).
• Alternative combinations are used if these variables deteriorate or unwanted drug effects occur. Antiretroviral resistance testing, both genetic (by searching viral RNA for sequences coding for resistance) and phenotypic (by testing antiretroviral agents against the patient’s virus in cell culture), also guide the choice of drug regimen, especially after virological failure.
• Pregnancy and breast feeding pose special problems. The objectives of therapy are to minimise drug toxicity to the fetus while reducing the maternal viral load and the catastrophic results of HIV transmission to the neonate. Prevention of maternal–fetal and maternal–infant spread is the most cost-effective way of using antiretroviral drugs in less developed countries. Maternal–fetal transmission rates are related to maternal viral load, with rates of 0.1% reported when maternal viral load is less than 50 copies per microlitre while on HAART. Where resources permit, access to safe alternatives to breast feeding should be provided to infected mothers.
• Combination antiretroviral therapy, especially the thymidine nucleoside analogue reverse transcriptase inhibitors zidovudine and stavudine, causes redistribution of body fat in some patients – the ‘lipodystrophy syndrome’. Protease inhibitors can disturb lipid and glucose metabolism to a degree that warrants a change to drugs with limited effects on lipid metabolism, e.g. ritonavir-boosted atazanavir, and the introduction of lipid-lowering agents.
• Impaired cell-mediated immunity leaves the host prey to opportunistic infections including: candidiasis, coccidioidomycosis, cryptosporidiosis, CMV disease, herpes simplex, histoplasmosis, Pneumocystis carinii pneumonia, toxoplasmosis and tuberculosis (often with multiply resistant organisms). Treatment of these conditions is referred to elsewhere in this text.2
• Improvement in immune function as a result of antiretroviral treatment may provoke an inflammatory reaction against residual opportunistic organisms (immune reconstitution inflammatory syndrome, IRIS). Although infrequent, this may present with development of new infections or worsening opportunistic infections, e.g. tuberculosis and cryptococcal disease.
• Antiretroviral drugs may also be used in combination to reduce the risks of infection with HIV from injuries, e.g. from HIV-contaminated needles and following sexual exposure to a high-risk partner. The decision to offer such post-exposure prophylaxis (PEP), and the optimal combination of drugs used, is a matter for experts; administration must begin within a few hours of exposure and continue for 28 days.
• Some drugs described here have found additional indications, or are used only, for therapy of non-HIV infections, e.g. adefovir for chronic hepatitis B infection.
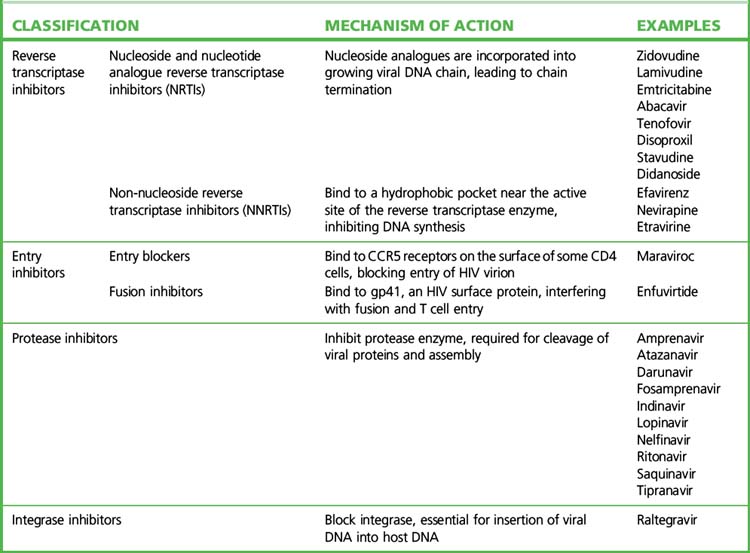
Nucleoside and nucleotide reverse transcriptase inhibitors
Protease inhibitors
• amprenavir, atazanavir, fosamprenavir (a prodrug of amprenavir), lopinavir, ritonavir, saquinavir, tipranavir, indinavir and darunavir.
Integrase inhibitors
Fixed-dose combinations of antiretroviral drugs
Fixed-dose combination antiretrovirals
| Combivir | zidovudine and lamivudine |
| Kivexa (Europe), Epzicom (USA) | abacavir and lamivudine |
| Truvada | tenofovir and emtricitabine |
| Trizivir | zidovudine and lamivudine and abacavir |
| Atripla | tenofovir and emtricitabine and efavirenz |
| Kaletra | lopinavir and ritonavir |
Influenza A
Zanamivir (Relenza)
• At-risk patients (those with chronic respiratory or cardiovascular disease, immunosuppression or diabetes mellitus, or over the age of 65 years).
• When virological surveillance in the community indicates that influenza virus is circulating.
• Only those presenting within 48 h of the onset of influenza-like symptoms.
Zanamivir retains activity against amantadine-resistant and some oseltamivir-resistant strains.
Cytomegalovirus
Foscarnet
Foscarnet finds use i.v. for CMV retinitis in patients with HIV infection when ganciclovir is contraindicated, and for aciclovir-resistant herpes simplex virus infection (see p. 213). It is generally less well tolerated than ganciclovir; adverse effects include renal toxicity (usually reversible), nausea and vomiting, neurological reactions and marrow suppression. Hypocalcaemia is seen especially when foscarnet is given with pentamidine, e.g. during treatment of multiple infections in patients with AIDS. Renal toxicity can be minimised with good hydration and dose modification. Foscarnet causes a contact dermatitis which can lead to unpleasant genital ulcerations due to high urine drug concentrations; this is potentially preventable with good urinary hygiene.
Drugs that modulate the host immune system
Interferons
Virus infection stimulates the production of protective glycoproteins (interferons) which act:
• directly on uninfected cells to induce enzymes that degrade viral RNA
• indirectly by stimulating the immune system
• to modify cell regulatory mechanisms and inhibit neoplastic growth.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


