Skeletal Hemangiomatosis/Lymphangiomatosis (Cystic Angiomatosis of Bone; Lymphangiectasis of Bone)
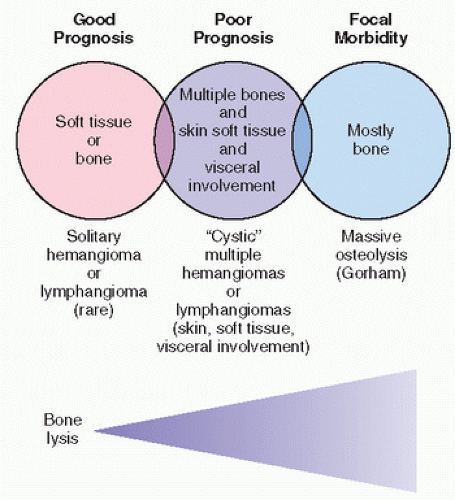
Systemic hemangiomatosis/lymphangiomatosis is a hamartomatous malformation involving the skeleton and often the visceral organs. It is usually diagnosed incidentally on x-ray films or after complications, which may include pathologic fracture and rarely soft tissue masses or even chylous or hemorrhagic effusion (
32,
35). Patients are usually in the first three decades of life at the time of diagnosis (
66). In childhood, the disorder may be asymptomatic until fracture occurs, in which case pain may be the presenting symptom. In adults, the symptoms may vary and include low back pain. Hemangiomas or lymphangiomas in the skeleton are often seen in association with visceral hemangiomas and lymphangiomas, which have a predilection for spleen, pleura, and skin.
Reid et al. (
34) reviewed 12 cases spanning four generations, referring to the entity as familial diffuse cystic angiomatosis of bone. He proposed autosomal dominant transmission. Affected persons were clinically asymptomatic, with normal laboratory values. Serial radiographs showed healing by sclerosis. A greater prevalence of this entity than heretofore clinically appreciated is now apparent.
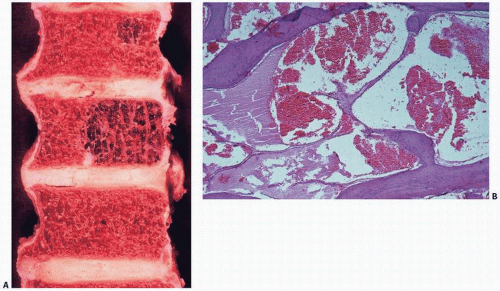
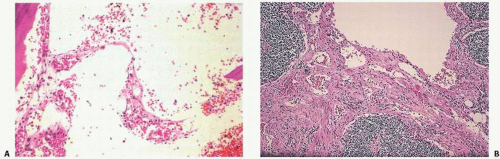
Roentgenographically, one notes multiple, cyst-like areas of bone destruction (
Fig. 11.10). There may be a fine peripheral rim of increased density. Isolated lesions may show the coarsened bone trabeculae of a solitary osseous hemangioma. Unlike the contour of bone involved by Paget disease, the contour of the exterior cortical bone is usually maintained in this condition, although vertebral collapse may occur (
Fig. 11.11). In rare cases, a blastic appearance predominates, mimicking metastatic cancer (
Fig. 11.12). Closer scrutiny, however, reveals central lucencies surrounded by dense, sclerotic bone. The lesions usually expand within the confines of the bone, but may involve the cortex, rarely breaking through the cortical shell.
Laboratory findings may be unremarkable, although increases in alkaline phosphatase activity have been noted. On gross examination, lesions are cystic and filled with a reddish fluid consistent with blood, or a lymphangiomatous fluid consistent with lymph origin. Often, there may be combinations of both lesions. Support for a lymph origin is noted in many cases that present with chylothorax or chylopericardium, and in many cases with a thymic mass seen in communication with lymph channels. Lymphangiograms may also show dramatic findings. Microscopically, the lesions consist of thin-walled cavities lined by flattened endothelial cells separated by collagen septa, filled either with blood in the case of hemangiomas or a proteinaceous eosinophilic material. These lesions are usually surrounded by normal or thickened lamellar bone trabeculae.
The prognosis in this disorder is variable, although usually it is self-limiting in its course. With extraskeletal involvement, however, the prognosis may be poor. The lesion is not hereditary, and most likely represents a maldeveloped vascular or lymphatic system.
Gorham Syndrome (Massive Osteolysis; Disappearing Bone Disease; Phantom Bone Disease)
Massive osteolysis was first described in 1838 by Jackson (
67), who documented a young man who had severe loss of bone of the upper extremity but lived to the age of 70 years, dying of unrelated causes. Numerous names have been used in the literature to describe this condition. In 1954, Gorham introduced the term massive osteolysis, and others have used the terms “disappearing” or “vanishing” bone disease and “phantom” bone (
54). In 1955, Gorham and Stout (
53) reviewed 24 cases, describing two additional cases, and concluded that the massive osteolysis was caused by a proliferation of vascular tissue, either vascular proliferation of blood or lymphatic tissue. Little has been added to the understanding or histologic assessment of this condition since that time. However, the identification of active osteoclastic resorption of bone and osteolysis in association with the proliferating vessels is now well documented. As reviewed by Choma et al. (
48), Gorham syndrome is best described as a clinical, roentgenographic, and histologic entity characterized by a nonfamilial proliferating vascular condition, most often originating in bone and causing lysis of all or part of the bone. The disease is characteristic in that it usually begins in one bone and spreads in a direct fashion to involve adjacent skeletal tissue. It may affect soft tissue by extension or be part of a marked, most likely developmental, abnormality of the lymphatic system. Identification of abnormal proliferating lymphatic tissue in visceral organs such as the liver, spleen, and lymph nodes supports this concept. Osseous involvement in reported cases shows a predilection for the mandible, ribs, scapula, humerus, and femur (
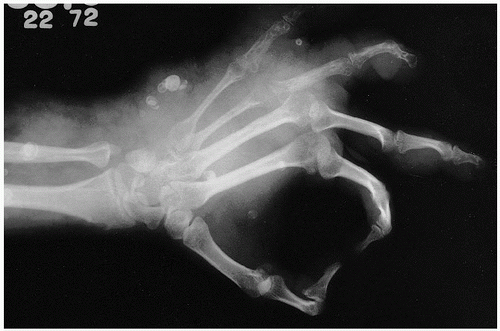
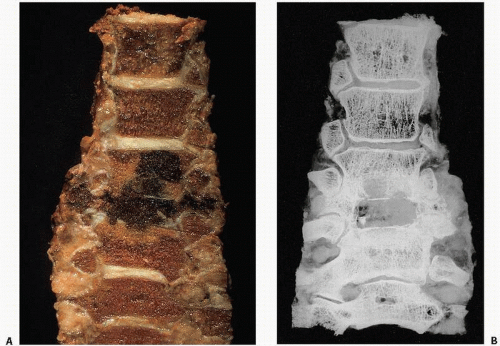
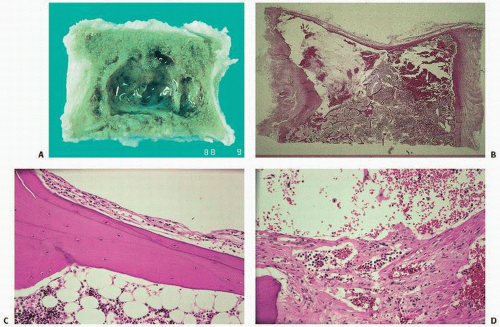
Fig. 11.13). Grossly, the bone is replaced by cyst-like vascular lesions (
Figs. 11.14 and
11.15).
Clinical presentation varies from the incidental finding of regional osteolysis to massive bone loss. Severe physical deformities, disabilities, and life-threatening complications may occur (
68). The onset is insidious, with dull pain in the affected region. Often, there is a history of preceding minor trauma or fracture in the remote past, but otherwise, the history is usually unremarkable. Localized pain as the chief complaint is frequent; however, a pathologic fracture or even deformity of the involved area with osteolysis may be found at initial presentation.
The age distribution of clinical presentation is varied. Presentation usually occurs before age 30 years, although the disease has been described in patients as young as 1 month to as old as 83 years. Gorham disease affects no particular sex or race, and does not appear to be associated with other disease entities or any pattern of genetic transmission. Bone lesions in Gorham disease appear to be equally distributed between the axial and appendicular skeleton; however, osteolysis has been described more frequently in the maxilla, mandible, clavicle, ribs, cervical vertebrae, pelvis, and femur. Osteolysis can be unifocal, with a single osteolytic focus affecting one bone area, or multifocal, with lesions occurring in more than one site simultaneously, particularly in a contiguous fashion.
The behavior of the osteolytic lesion is varied, ranging from slow, asymptomatic progression with spontaneous arrest after a number of years (
69) to an extremely rapid and aggressive bone resorption that causes disability and even death when vital organs are involved.
Radiographic changes occurring in massive osteolysis include initial intramedullary and subcortical radiolucent foci described as a “nondescript patchy osteoporosis” (
57). Concentric shrinkage is characteristically seen in tubular bones, with tapering of the involved ends. Complete resorption of the involved bone follows, barring spontaneous arrest (
Fig. 11.16). The osteolytic process may progress from intraosseous to extraosseous, with involvement of contiguous bones. Torg and Steel (
59) illustrated the sequential radiographic changes occurring during a 10-year period in the hip of an 11-year-old with massive osteolysis. A diffuse lytic lesion within the confines of normal bone architecture was followed by increasing bone loss and deformity spreading to adjacent structures and across joints, and concentric shrinkage of involved long bone.
Arteriography, venography, and lymphangiography most commonly give only indirect evidence of Gorham disease and are not considered useful. Results of technetium pyrophosphate bone scans are routinely negative, or they demonstrate decreased uptake, confirming the absence of mineralization activity, or new bone formation or osteoblastic activity histopathologically. MRI has revealed a high signal intensity on T2-weighted images. There is no useful laboratory test to diagnose the disorder. The histopathology of Gorham disease is characterized by nonneoplastic, thin-walled lymphatic or blood vessels. There is usually little inflammation. One may observe mononucleated clear cells and multinucleated osteoclast-like cells.
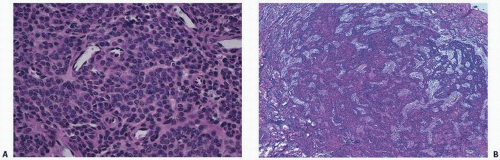
The replacement of the bone by proliferating vascular tissue is often seen in association with fibrous connective tissue. The vascular tissue varies from proliferating small capillary-type vascular tissue to that of proliferating or large hamartoma-like lymphatic- and/or endothelial-lined vascular channels (
Fig. 11.14). Although the tissue is always histologically benign, the proliferation of vascular channels permeates the bone, cortex, and soft tissue in an aggressive fashion, similar to the uncontained proliferation of synovium in rheumatoid arthritis. There is usually little inflammation, although transient inflammation occurs, and fibrosis has been described in long-standing lesions. There are now several well-documented cases showing active mononuclear cells or phagocytic cells or even osteoclast-like cells resorbing bone at the membrane margin of the proliferating vascular channels (
Fig. 11.15).
The prognosis in Gorham syndrome varies considerably. There is usually minimal disability, and the disease usually spontaneously regresses. However, with involvement of vital structures, such as the lung, death may ensue. Choma et al. (
48) reported that 38 percent of patients with spinal and/or chest wall lesions died as a result of their disease, usually from extensive chylothorax. The development of chylothorax is particularly foreboding in that it may be very difficult to control the production of chyle.
Numerous therapeutic modalities have been described, including radiotherapy, bone grafts, prosthesis implantation, and (for chylothorax) irradiation and pleural adhesion therapies with minocycline and bleomycin (
70). Although bone grafts may be resorbed in this condition, success with bone engraftment as well as prosthesis implantation has been reported. The condition often
spontaneously remits, and improvement and regrowth of bone may be seen, and in very rare instances, complete reconstitution of the bone.
Radiotherapy has been used with reputed success for massive osteolysis of the chest with hemothorax (
71). Radiation doses of less than 20 Gy are generally ineffective. Arrest of osteolysis has been reported with doses of 25 to 60 Gy.
Numerous etiologies have been suggested for Gorham disease, including posttraumatic hyperemia, similar to mechanisms suggested for bone resorption and reflex sympathetic dystrophy. The proliferation of fibrovascular tissue uncontrollably resorbing bone is similar to that seen in rheumatoid synovium. More than likely, the production and proliferation of vascular tissue are somehow directly linked to osteoclast activation or other bone-resorbing factors. Several mechanisms may be at work in this association. Endothelial cells may directly mediate bone resorption or modulate the extracellular matrix by producing enzymes that can degrade the matrix (
72).
Because the regulation of cell growth most likely involves the interaction between growth factors and matrix proteins, the former (such as transforming growth factors) having great influence on cells of vascular lining tissue, and the latter (such as collagen I, III, IV, and V, elastin, and osteopontin) being abundant in the extracellular compartment of blood vessels, the potential for vascular lesions to remodel bone is great (
72).
It has also been postulated that mesenchymal cells, which accompany blood vessels, may act as progenitor cells for lineage 2 osteoclasts. It has been observed that circulating monocytes in tissue culture, for example, are capable of inducing bone resorption.
Because no abnormality of the parathyroid glands is discernible in this condition, nor any systemic evidence of osteoclast-induced hypercalcemia via a parathormone mechanism, osteolysis appears to be caused by local factors and most probably is directly linked to the proliferating vascular channels.
A plausible explanation for the osteolysis in Gorham disease is that the proliferation of lymphatic endothelial cells (LECs) and blood endothelial cells (BECs) is stimulated by increased levels of vasoactive endothelial growth factors C and D (VEGF-C and VEGF-D) and macrophages-derived VEGF-A. In addition to the VEGFs, macrophages (progenitors of osteoclasts) can produce interleukin-6, all stimulators of osteoclast differentiation and all shown to be increased in the serum in different phases of the diseases (
68). In fact, the endothelium of lymphatic vessels has been shown to harbor toll-like receptors that can upregulate osteoclast activation via tumor necrosis factor alpha and interleukin-6 (
73). In addition, osteoblast progenitor cells may be inhibited by LECs and factors produced by osteocytes like sclerostin, Dickkopf- and soluble frizzled-related proteins.
The Lymphatic “Chylous” Variant of Gorham Disease
Gorham disease may also produce an indolent lymphatic proliferation that, over time, creates excessive chylous fluid (
74).
Documented abnormalities of the lymphatic system including lymphangiomatous masses, lymphangiectatic dilations (small bowel, spleen, pancreas, thymus, and bones), and absence of a
portion of the thoracic duct have been shown. These findings are important because therapeutic attempts focusing on identification and ligation of the duct may be futile, as has been reported. Podevin et al. (
75) recognized this futility by using the phrase “thoracic lymphatic dysplasia not further defined.”
Dilated lymphatic channels (lymphangiectasia) or accumulation of lymphangiomatous tissues in the pleural and peritoneal tissue, the diaphragm, mediastinum, and spleen correspond to known anatomic tracking of the thoracic duct.
There were several common findings during autopsies of five patients with Gorham disease (
Table 11.4) (
54,
55,
56,
74,
75,
76,
77). All had abnormal vascular proliferation that involved bone sites, some described as lymphatic and some admixed with fibrosis. Sites of involvement include pleural tissue, spleen, nodes, and vertebrae, as in our case. There is a predilection for osseous involvement of the clavicle, scapula, ribs, and vertebrae, as revealed by autopsy or premortem imaging. Numerous patients died from chylous effusions and pneumonia.
The lymphatic variant of Gorham disease may be a distinct variant of the disease. Some have suggested that Gorham is a generalized disorder of lymphangiogenesis and should be distinguished from cases that are a generalized lymphatic abnormality involving bone (
78). Approximately 17 percent of patients with Gorham disease have chylothoraces develop, and approximately one-half die. Gorham disease generally affects younger patients and seems to have no racial or gender predilection, and most osseous involvement occurs in the shoulder region and vertebrae. In our patients and in reported cases, chylous effusions were frustrating to treat, with a poor prognosis.
A distinct variant of Gorham disease has been proposed on the basis of autopsy findings (
Table 11.5). A maldeveloped (or totally absent) thoracic duct may lead to the development of an aberrant collateral lymphatic circulation including dilated lymphatic channels (lymphangiectasia) and lymph-rich soft tissue masses (lymphangiomas). Collateral lymphatic circulation develops and extends to the nascent intraosseous lymphatic system. This bone lymphatic tissue contains activated endothelial lymphatic cells leading to increased activity of osteoclasts, which lead to marked osteolysis.
Although there is no consensus regarding treatment of Gorham disease, therapeutic attempts must focus on the morbid and deadly chylous effusions. Surgical interventions in patients with Gorham disease complicated by chylous effusions have been mostly unsuccessful including pleurectomy, pleurodesis, thoracentesis, and thoracic duct embolization and ligation (
76).
Thoracic duct ligation does not seem to be anatomically sound in all cases. The lymphatic dysplasia must be localized by imaging studies and addressed regionally. Although this is a challenging and elusive goal, patients with regionalized lymphatic dysplasia can be treated by an aggressive therapy such as radiotherapy or specific excision of the lesion. Osteoclastic resorption needs to be addressed in this condition, and may best be blocked by blunting local tissue factors or by using antiosteoclastic agents such as new-generation bisphosphonates.
Applying agents that interfere with osteoclast stimulation has been attempted in this condition. Agents previously utilized for antiosteoclastic activity include bisphosphonates, interferon alpha 2b, anti-VEGF-A antibody (Bevacizumab), propranolol, low molecular weight heparin, steroids, vitamin D, and calcitonin (
68). Since the etiology of Gorham remains enigmatic, therapy is still frustrating.
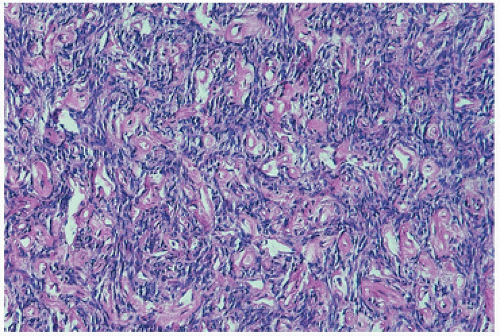
Vascular tumors of bone are rare and vary from well-differentiated and locally aggressive tumors (epithelioid hemangiomas, previously called low-grade angiosarcomas) to highly anaplastic, poorly differentiated metastasizing neoplasms (angiosarcomas). Nomenclature is confusing, reflecting current debates over prognostic significance. Surgeons are often frustrated by the inability of pathologists to provide better guidance in the natural history of these lesions.
For those interested in the passion that nomenclature bias can invite, the letters published in the
American Journal of Surgical Pathology regarding terminology of these lesions are worth reading (
79,
80,
81,
82). In general, there is a gradient of aggressive behavior with increasing pleomorphism and atypical mitotic activity, but lesions, especially multicentric lesions, may be hard to predict (
83).
Peculiar features of vascular tumors of bone include concomitant lesions of soft tissue and multicentricity. The often-observed multifocality of these lesions remains enigmatic with regard to prognosis and therapy. Multiple primary tumors may be mistakenly confused with metastatic disease. The phenomenon of multicentricity in these lesions, often with both visceral and bone involvement, suggests corollaries with the KS phenomenon.
Concomitant histologic foci of solid spindle cell regions can be confused with other spindle cell lesions.