(1)
NIDDK, National Institutes of Health, Bethesda, MD, USA
(2)
NHLBI, National Institutes of Health, Bethesda, MD, USA
Abstract
As the other chapters attest, sensitivity of fluorescent molecules to their local environment has created powerful tools in the study of molecular biology, particularly in the study of protein, DNA, and lipid dynamics. Surprisingly, even events faster than the nanosecond lifetimes of fluorophores are important in protein function, and in particular, events lasting just a few ps reflect on water motion and the coupled dynamics of proteins. These ultrafast phenomena can best be studied by using the same laser that excites fluorescence to also “strobe” the emission, providing sub-picosecond time slices of the action. We explain the strobing “upconversion” technique and some limits on its execution.
Key words
Time-resolved fluorescenceUpconversionTryptophanProtein dynamics and photodamage1 Introduction
1.1 A Brief History of Upconversion Spectroscopy
The newly discovered red ruby laser was focused into quartz in 1961 by Franken et al. [1], and the output beam was passed through a prism spectrograph, allowing them to observe a much weaker 347 nm (doubled frequency) spot. While frequency mixing of many types was known in microwave circuits, RF cavities, and (of course) low-frequency electronics (esp. household radio heterodyne circuit stages), optical mixing had not previously been seen.
The other nonlinear optical phenomena followed, including higher harmonic generation and generation of both optical difference frequency (e.g., for novel monochromatic IR beams) and sum frequency generation. The latter, conceptually identical to second harmonic generation, gained favor as a method to convert weak infrared signals into visible signals amenable to detection with more conventional cameras and detectors. The redder signal was said to have been frequency “upconverted” into the visible.
To accomplish upconversion, a stronger (and usually redder) optical field was needed to mix with the weak signal. Just as in electronics, mixing demands the voltages be high enough to elicit a distorted (nonlinear) response of the mixer material to the sinusoidal waves. Otherwise, the beams simply superimpose in the crystal, and the electrons simply track the superposition of electric fields. The strong electric fields of short laser pulses were just what was needed to reveal second-order crystal nonlinearity and mixing. Of course, to create mixing, the electric fields had to superimpose not only in space, but also in time. This created the opportunity to auto or cross-correlate laser pulses, since the mixed signal output envelope is a temporal convolution of the input pulses.
Autocorrelation and cross-correlation of pulses set the stage for not only transforming wavelengths, but also quantifying ultrafast kinetics. The photocathodes all found at the heart of photomultipliers, vacuum diodes, and streak cameras invariably produce a spread of photoelectron ejection energies in response to a laser pulse, meaning those electrons take slightly different paths (and times) to their eventual detection. As a result, phenomena faster than several ps were intractable for even the fastest photodetectors. Optical mixing was the best solution.
In chemistry, the first published use of upconversion to trace a luminescent process was by Halliday and Topp [2]. The method spread rapidly in photophysics, as briefly reviewed by Barbara and colleagues [3].
Ultrafast spectroscopy (particularly time-resolved fluorescence) has been a fruitful area of biological research. While it is, at first, counterintuitive to think that nanosecond (or worse, picosecond and faster) events bear relevance to enzymatic and structural changes taking a billion times longer, the facts have been clear: changes in the fluorescent signals on or in proteins, membranes, and DNA give intimate details about the size, flexibility, fluidity, and interrelationships (including proximity and motion) of these various macromolecules. Several other chapters in this book will provide examples of this windfall.
Among the various biophysical targets of interest, upconversion spectroscopy quickly became most popular in a field that, unsurprisingly, is hungriest for time-resolved data tracing the flow of photonic energy: Photosynthesis. The study of photosynthetic pigments and their photochemical and functional physical arrangements includes relevant kinetics on everything from femtosecond through minute timescales [4–6].
1.2 Use of Upconversion to Study Proteins
Tryptophan is both the blessing and curse of protein dynamics spectroscopy. Present in most proteins, yet sparse enough that most have only a few Trp (W) in their sequence, it represents a local reporter that is intricately linked to its surroundings as it performs its native structural and/or electronic duties [7]. Trp has been studied for years with time-resolved spectroscopy on the nanosecond scale, and the best software and genetic manipulations have been applied to the task of isolating the signals belonging to single Trps within proteins [8, 9]. Such signals tell us about local mobility and folding, overall tumbling of the protein, proximity to FRET acceptors such as NADH [10] or heme [11], and the availability of the Trp ring to solvent quenchers [12].
Single Trp in proteins often provides multiexponential decay behavior, a fact that had led to a long-standing controversy about why the protein environment is not homogeneous on that timescale. Two camps evolved, one (observing comparable features of indole in glycerol) and seeing Trp in proteins also tended to have longer decays at the red side of the spectrum, invoked ns solvent relaxation [13, 14]. A second camp invoked Occam’s razor to point out the decay surface was compatible with simple heterogeneity—perhaps by mechanisms as simple as side-chain rotamers (or more broadly, conformers) [15, 16]. The nanosecond fluorometry “heterogeneity vs. relaxation” debate went on throughout the last three decades, and the combination of studies on crystals (where orientation modified preexponential weights, a result incompatible with relaxation) [17] and studies on constrained Trp derivatives [18] pushed toward a consensus that on the nanosecond scale, Trp decay in proteins was often dominated by heterogeneity.
This was not to say relaxation was absent; instead, it was suggested that it was mostly complete within ~100 ps (the lower time resolution typical of nanosecond instruments). Most telling was the absence of a negative preexponential term on the red side of the Trp emission, a signal known to flag relaxation (or any other excited state reaction) [19]. Advocates of relaxation models, while agreeing this rising term should be present, rightly argued that the negative preexponential could be quite small and therefore easily lost in either noise or the contributions of short-lived contaminants (perhaps photoproducts) [20].
A progression of nanosecond lifetimes associated with a progression to the red in spectral position (redder “DAS” correlating with longer lifetime) was also suggested as reflective of relaxation, and the paucity of proteins breaking that pattern was noted. Only recently, the intrinsic nature of the nanoheterogeneous quenching mechanism has been found to favor this correlation [21].
Early picosecond studies of Trp, meanwhile, were carried out laser-based chemical physics community, and they were generally remote from the nanosecond Trp controversy. Treating Trp like other dyes (e.g., coumarins) in solvents, they obtained time-dependent fluorescence Stokes shifts (TDFSS) (or fluorescence dynamic Stokes shift, FDSS) and deduced correlation functions, concerned primarily with the spectral movement rather than deactivation [22, 23]. Exceptions to this focus were studies of Trp in heme-containing proteins, where efficient FRET was anticipated from Trp to Soret bands, leading to sub -30 ps overall lifetimes [24, 25]. The early behavior of Trp was also probed for emission anisotropy [26], where, unfortunately, some Raman contamination to curves (apparently) made Trp appear to be a complex, vibrational-cooling dominated system (likely too complex to be useful in ps motion studies). In the late 1990s, however, Shen et al. [27] showed that Trp not only produced strong 1.6 ps solvent relaxation spectra in water, but also had simple anisotropy behavior; oscillators within Trp analogs were seen to perform internal conversion (reaching stable orientation) well within 100 fs. This opened the door to exploiting Trp without the complexity concerns previously mentioned.
A recurrent theme in protein dynamics is the protein–water interface. Data available from X-ray to neutron diffraction point to an ordered water layer in direct contact with the protein. The dynamic properties of this ordered layer of water are not clear, and if they are different it poses a question: Are these water molecules simply slaved to the protein charge distribution and dynamics or does the ordered water also affect protein dynamics? The nuclear overhauser effect (NOE) allows the investigator to probe water relaxation and compare it to bulk water with sub-nanosecond time resolution; further, nuclear magnetic relaxation dispersion (NMRD) has shorter effective time resolution. FDSS, using fluorescence upconversion on Trp residues of Monellin, showed spectral downshifting that was slower compared to those observed for single indole molecules in solution. It was then proposed that such a slow (8–20× slower) shift was the signature of a layer of organized water (also called “biological water”). This new “slow” water on protein would have solvation dynamics in the range of 30 to hundreds of picoseconds, in contrast to the few picoseconds measured for bulk water. In protein dynamics, X-ray diffraction can provide initial structures that, along with MD simulation, can predict stepwise information on this layer of solvation water. Recent results have also been gleaned from 17O spin relaxation, providing values for the dynamics of water at the interface that are consistently faster that those inferred using FDSS.
When the laboratory of laureate A. Zewail published relaxation profiles for Trp within the sweet protein Monellin, finding not only the 1.6 ps “bulk water” relaxation term but also a redshift that required 10s of ps, they (as mentioned above) attributed this slower process to exchanging water between bulk and bound sites with off-rates near 20 ps [28]. Importantly, they also made emission anisotropy measurements in a variety of protein states, confirming the viability of Trp as a probe of segmental motion [29, 30].
Noting that the raw TRES (Time Resolved Emission Spectra) of Monellin did not rise after a few ps, Xu et al. [45] showed that this 20 ps behavior of Monellin was actually dominated by an ultrafast quenching process (the blue side was simply being consumed more rapidly than red) rather than a conservative steady redshift like that seen in bulk water. In other words, no negative preexponential was found for the 20 ps term. This essentially caused the two model controversy from ns Trp studies to reemerge at 100× faster rates.
Other proteins [31, 32] clearly did show the negative amplitude feature. Was relaxation always present and was it simply “masked” in some proteins by superposed quenching? Was it unique to proteins with particular folds, partial unfolding, or denaturation? Monellin in denaturants was certainly a strong exhibitor of ~50 ps relaxation, but the presence of a viscous denaturant complicates any dynamics gleaned from that picture [28].
One approach to the “masking” question was mutagenesis—by removing likely quenchers in the neighborhood of Trp (and replacing them with benign side chains), a quencher-free relaxation was seen [33].
Meanwhile, the rapid quenching, naturally unseen in nanosecond instrumentation, was termed QSSQ (Quasi-Static Self Quenching) in accordance with Chen et al. and it was found to be effective even in simple dipeptides [34, 35]. More importantly, it was seen that there were not only < 100 ps quenching rates, but also losses faster than the instrument response of 0.2 ps (200 fs). The “initial brightness” (taken typically at ~1 ps) could be used to sense this, and the sum of these ultrafast terms reconciled yield defects in a variety of peptides [36]. An actual biological use for QSSQ was found in the eye lens proteins termed crystallins; apparently, animals have evolved to keep these proteins from oxidizing in ultraviolet light by creating very strongly quenched Trp environments [37]. Thus, the ultrafast QSSQ is a useful probe of proteins independent of the relaxation phenomenon it masks.
1.3 Molecular Dynamics (MD) as a Tool to Decode Protein Dynamics
Molecular dynamics (MD) simulations have contributed in shaping today’s awareness of proteins acting as dynamic complex systems, and they also provide a unique window on the behavior of protein and solvation water in the femto- to picosecond timescale.
Attempts to follow protein dynamics involving the use of covalently bound extrinsic fluorescence probes produced mixed results. In some cases it was difficult to deconvolute the protein internal dynamics from those of the dye [38]. The protein dynamics community pursued many different routes. Alongside fluorescence dynamic Stokes shift (FDSS), a number of different techniques were employed to probe protein dynamics, esp. NMR and nuclear overhauser effect (NOE) with MD simulations performed in parallel [39–42].
Decoding of the Trp fluorescence signal and its correlation to short- and long-range molecular dynamics in proteins is not an easy task. MD simulations proved to be essential in helping with the data interpretation; hybrid QM-MM simulation methods have indeed been successful in predicting fluorescence emission maxima and quantum yields for Trp residues in a variety of proteins [43, 44]. Importantly, the study done on γS and γD-crystallin revealed multiexponential decays in the ps time domain along with a very rapidly quenched population, and QM-MM simulations done on such residues clearly disclosed that nearby charges were the cause for the fast quenching: A Trp charge transfer (CT) state was produced with the amide backbone, stabilized by nearby water and charged residues; thus, the predicted Trp fluorescence lifetime was drastically reduced [37]. As mentioned above, nature might have selected such “enhanced” quenching mechanisms in crystallin to insure longevity in an eye lens proteins heavily exposed to UV light.
The most recent work in this area by our group concludes that relaxation is ubiquitous but can be masked by the equally valuable QSSQ phenomenon. We find relaxation in GB1 is remarkably refractory to pH-induced changes in protein structure that have profound effects on ns emission [45]. Further, substitution of Trp analogs via auxotrophic biosynthesis can eliminate the QSSQ term. For example, relaxation within the aforementioned Monellin protein (dominated by 20 ps quenching) is “unmasked” when 5FTrp replaces Trp [46].
Upconversion of Trp then gives us access to two different valuable indicators of protein dynamics: relaxation, with wider extent, and QSSQ, with precise dependence on Trp nearest neighbors.
2 Materials
2.1 Upconversion Spectrophoto fluorometer and Experimental Instructions
Fluorescence upconversion is a derivative of the nonlinear pump probe technique, and a spectrophotofluorometer was developed especially for studying tryptophan fluorescence dynamics in proteins, which is shown in Fig. 1. A ~400 mW pulse train with a typical pulse duration of 120 fs and a repetition rate of 82 MHz was generated from a mode-locked Ti: sapphire laser (Tsunami, Spectra-Physics) pumped by a diode-pumped solid state (DPSS) laser with the power of 5 W (Millennia, Spectra-Physics). It seeds a Ti: sapphire regenerative amplifier (Spitfire, Spectra Physics) pumped by a diode-pumped nanosecond green laser with average power of 15 W at 5 kHz (Empower, Spectra-Physics). The output amplified pulses at 885 nm typically had a pulse energy of 0.2 mJ and an autocorrelation pulse width of 350 fs (see Fig. 2) at a repetition rate of 5 kHz (i.e., ~1 W output power). Ultraviolet excitation pulses at 295 nm with an average power up to 10 mW were obtained from nonlinear harmonic generation (a 1 mm BBO crystal and 0.5 mm BBO crystal for doubling and tripling, respectively). This UV beam was separated from the infrared beam (fundamental) and visible (doubled) by two dichroic mirrors and by spatial filtering (using a pinhole). Power should be carefully attenuated to below 1 mW before excitation of the sample to avoid photodegradation, hole burning, and other undesirable effects. A zero-order half wavelength plate (HWP) was put on a computer-controlled servo stage and used to change the polarization of the UV beam. A circular array of thin cells (T-20, NSG Precision Cells) with a path length of 1 mm in a delrin stacked slotted disk was used to hold samples, and the total volume was about 2 ml. The disk was spun continuously (up to 5 m/s circumferential motion) to make each UV pulse excite a slightly different sample spot. The remaining infrared fundamental pulse was reflected from a broadband hollow retroreflector on a computer-controlled precision stage and used as a gate pulse for the upconversion process. The fluorescence emission was collected by parabolic mirrors, passed through a long pass filter (F), and focused into a 0.5 mm thick BBO mixing crystal. The upconversion signal was produced via type I sum frequency generation with the gate pulse in the crystal. Here the combination of a thin BBO crystal and mirrors is selected for reducing the limitations of time resolution imposed by group velocity mismatch and dispersion. The time evolution of fluorescence at any given wavelength can be obtained by using a computer to set the BBO crystal angle, monochromator wavelength (at ω F + ω p), and scan delay stage. The delay stage is comprised of a hollow cube corner retroreflector (Newport UBBR1-5I) mounted on a cross roller slide (Parker) linear stage driven by compumotor motor, combined with appropriate NIR relay mirrors.
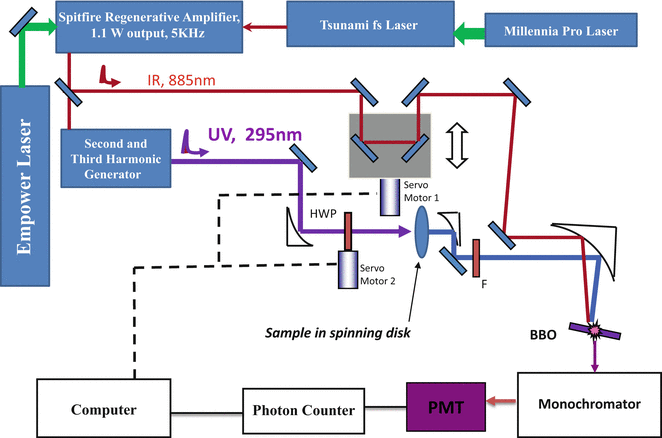
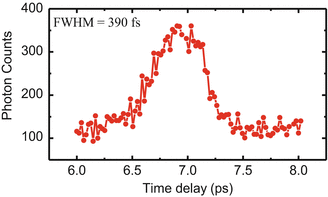
Fig. 1
A layout of the upconversion spectrometer setup
Fig. 2
A typical snapshot of the response time profile of upconversion spectrophotofluorometer
To reject the strong background signals (infrared laser and its second harmonic generation, remnant UV, and unconverted fluorescence) accompanying the upconverted signal, a non-configuration was arranged between infrared probe and fluorescence. The use of a monochromator helps in reducing these unwanted signals at the PMT, also improving the spectral bandwidth. A double monochromator may be necessary if this problem is severe. Polarizations of gated fluorescence were determined by the orientation of nonlinear crystals, so no extra linear polarizer was needed (for anisotropy calculation, the grating factor G ~ 1). By angle tuning the mixing crystal, the upconverted fluorescence signal, with a wavelength in the range 230–280 nm, always polarized in the same direction, was directed into a monochromator (Triax 320, Jobin Yvon Inc. with a bandwidth of 0.5 nm) and a solar blind photomultiplier tube (R2078, Hamamatsu, dark rate <1cps). Amplified SBPMT signals were discriminated and then recorded by a gated single photon counter (994, EG&G Ortec). Photon arrival events were held to less than 5 % of the repetition rate to minimize “pileup” (which might flatten transient). One should mention that there are two main sources of noise for the detection. If the signal is sufficiently greater than the PMT dark current, the power fluctuations of the infrared laser beam may be the primary noise source. If the signal is very low, the shot noise from the dark current of the PMT will be the limiting factor, and in this case, cooling the PMT is advisable.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


