UK Guidance on Wholesale Distribution Practice
The Application and Inspection Process “What to Expect”
Applicants for a new wholesale dealer’s licence (WDA(H)) or existing licence holders wishing to vary their licence should apply using the MHRA Process Licensing Portal accessible via the MHRA website.1
MHRA acting as the licensing authority will only issue a wholesale dealer’s licence when it is satisfied, following an inspection of the site(s), that the information contained in the application is accurate and in compliance with the requirements of the legislation.
When appropriate, MHRA may refuse to grant a wholesale dealer’s licence or may grant a wholesale dealer’s licence otherwise than as applied for. In such cases the licensing authority will notify the applicant of its proposals. The notification will set out the reasons for its proposals and give the applicant a period of not less than 28 days to respond.
Planning
Fee bearing inspections of licensed wholesale dealers are carried out to assess the degree of compliance to standards of Good Distribution Practice (GDP) and compliance with the provisions of the licence.
Inspections of wholesaler dealers (WDA(H) holders) are undertaken as part of the risk-based inspection programme, further details of which can be found in another section of this guide.
Notification
Advance notice of inspection is normally given to a company, unless circumstances require that an unannounced inspection should take place. The timing of the inspection would normally be notified in writing by the inspector.
In accordance with the GDP risk-based inspection process, sites will be required to complete a Compliance Report in advance of inspection. Further information and guidance can be found in our risk-based inspections section.
Conduct
The major stages of the inspection process are:
● the introductory or opening meeting
● the detailed site inspection
● the summary or closing meeting
Introductory or opening meeting
The purpose of the meeting is for the Inspector to meet with the appropriate key personnel from the company to discuss the arrangements for the inspection. The Inspector would typically confirm the nature of the business, premises and security arrangements, areas to be visited and any documentation which may be required.
Site inspection
The purpose of the site inspection is to determine the degree of conformity of the operations to the requirements of Good Distribution Practice and to assess compliance with the terms and conditions of licences issued under the appropriate legislation or with details submitted in support of an application for a licence. The inspection will typically involve visits to goods receipt, storage and dispatch areas (including ambient and refrigerated), returns/quarantine area, interviews with key personnel and a review of stock movement and quality system documentation including product recalls. Any observations, recommendations and deficiencies noted during the inspection would normally be discussed with the company representatives at the time.
During inspections of manufacturing and wholesale operations, samples of starting materials, work in progress and finished products may be taken for testing if an Inspector considers that this might assist in the detection of quality deficiencies. Occasionally, samples may be taken when these cannot be obtained from other sources, for routine surveillance purposes.
Summary or closing meeting
The purpose of the meeting is for the Inspector to provide the company with a verbal summary of the inspection findings and to allow the company to correct at this stage any misconceptions. The Inspector would typically summarise the definition and classification of deficiencies they propose to report and the company are encouraged to give an undertaking to resolve the deficiencies and to agree a provisional timetable for corrective action. The Inspector would also describe the arrangements for the formal notification of the deficiencies to the company (the post-inspection letter) and what is expected as a response.
Deficiencies are classified as follows:
● Critical deficiency:
Any departure from Guidelines on Good Distribution Practice resulting in a medicinal product causing a significant risk to the patient and public health. This includes an activity increasing the risk of falsified medicines reaching the patients. A combination of a number of major deficiencies that indicates a serious systems failure. An example of a critical deficiency could be:
Purchase from or supply of medicinal products to a non-authorised person; Storage of products requiring refrigeration at ambient temperatures; Rejected or recalled products found in sellable stock.
● Major Deficiency:
A non-critical deficiency: which indicates a major deviation from Good Distribution Practice; or which has caused or may cause a medicinal product not to comply with its marketing authorisation in particular its storage and transport conditions; or which indicates a major deviation from the terms and provisions of the wholesale distribution authorisation; or a combination of several other deficiencies, none of which on their own may be major, but which may together represent a major deficiency.
● Other Deficiency:
A deficiency which cannot be classified as either critical or major, but which indicates a departure from Guidelines on Good Distribution Practice.
The choice of company representatives at the meeting is primarily for the company to decide, but should normally include the senior staff who were present during the inspection and the Responsible Person (RP).
Depending upon the inspection findings and the response from the company during and following the inspection, the Inspector may take one of a number of actions ranging from:
● issuing a GDP certificate confirming essential compliance with GDP
● referral to the Compliance escalation process or the Inspection Action Group (IAG) for consideration for adverse licensing action where serious non-compliance is found.
Further information on the Compliance escalation process and IAG can be found in this publication.
Company responses
The inspected site is expected to provide a written response (by letter or email) to the post-inspection letter within the required timeframe. The response should consider the context of the deficiency within the overall quality system rather than just the specific issue identified. The response should include proposals for dealing with the deficiencies, together with a timetable for their implementation. The response should be structured as follows:
● Restate the deficiency number and the deficiency as written below.
● State the proposed corrective action.
● State the proposed target date for the completion of the corrective action(s).
● Include any comment the company considers appropriate.
● Provide evidence supporting any corrective action where it is considered appropriate.
Inspection report
Once the Inspector is satisfied that any necessary remedial action has been taken or is in hand and that the site is essentially in compliance with GDP, an inspection report and GDP certificate are finalised.
Risk-based Inspection Programme
Introduction
MHRA has been incorporating elements of risk management into its inspection programme for a number of years. A formal risk-based inspection (RBI) programme was implemented on 1 April 2009, following public consultation MLX 345. The RBI programme covers all aspects of good practices associated with the inspection of clinical, pre-clinical and quality control laboratories, clinical trials, manufacturers, wholesalers and pharmacovigilance systems. The primary aim of the RBI programme is to enable inspectorate resources to focus on areas that maximise protection of public health while reducing the overall administrative and economic burden to stakeholders.
Sentinel risk information module
Working with technology partners Accenture, MHRA established its Sentinel IT system in 2005 which is used by most agency business areas to manage business processes for:
● marketing authorisations
● pharmacovigilance
● clinical trials
● manufacturer’s and wholesale dealer’s licences
● inspections
● issuing GMP and GDP certificates and automatic loading of these into the EMA’s EudraGMDP database.
In February 2013 a newly developed Sentinel risk information module was deployed to expand upon the paper-based RBI system initiated in 2009.
The Risk Estimation Tool uses the intelligence data collected on regulated companies, their respective sites and previous inspection results across all GxP areas to predict a risk score as “likely next inspection result”. This score is calculated for every site and can be interpreted as a weighted sum of inspection findings. Companies/sites are ranked based on predicted risk and business rules are applied to suggest a next inspection date.
A planning step allows inspectors to accept or reject the suggested date taking into account other information which may not be included in the statistical calculation. For estimation of the risk score, the tool uses a logistic regression statistical model incorporating all data elements for all companies and sites. The model is fit (i.e. recomputed) monthly based on the most recent data extracted from Sentinel. The Empirica algorithm software was designed by Oracle Health Services to provide detailed analysis of the risk information. MHRA first used Empirica software in 2006 for pharmacovigilance signal detection and management.
The model estimates the association between inspection findings and other covariates (events) observed in data. The algorithm makes a global estimate on how these events affect inspection score within a GxP and then applies this when these events are recorded in the future. As a result those factors which are statistically most relevant to risk will receive the highest weighting and this will be continuously updated as more events are recorded. The model looks at events over a five year period but applies greater significance to more recent data.
Current implementation status
A number of aspects of the algorithm are being validated including;
● The risk score
● The weighting of inspection outcomes
● The weighting of the risk events
● The generation of proposed inspection dates from the risk score
● Inclusion of all appropriate risk events
The algorithm is being assessed on an individual GxP basis as well as across the GxPs. The algorithm output is being compared against the existing RBI processes within the GxPs. Until the algorithm has been successfully validated the existing risk-based inspection scheduling processes will remain in place.
GDP risk-based inspection (RBI) programme
The GDP risk-based inspection process commenced for all wholesale dealer’s licence holders on 1 April 2009.
Compliance report
Sites will be required to complete a Compliance Report in advance of inspection, this will be prompted by the inspector. Guidance to completing the report can be found within the document. The Compliance Report should be returned to your inspector prior to the inspection.
Risk rating process
Inspectors use the inspection outputs along with a number of other factors to identify a risk rating for the site, which equates to a future inspection frequency. As this process is not concluded until the inspection is closed the risk ratings will not be discussed at the closing meetings. However a copy of the full inspection report which includes the full risk rating rationale is provided to sites once the inspection has been closed.
Issue of a certificate of GDP compliance and/or support of the site on the relevant licence is indication of meeting the minimum level of GDP compliance. Risk ratings identify the degree of surveillance required within the licensing and inspection program. There is no intention that sites be rated against each other as a result of risk ratings assigned by MHRA. Risk ratings can change following inspection resulting in either increased or decreased risk. Inspection risk ratings will not be published by MHRA.
There will be no formal process of appeal against risk ratings and future inspection frequency. However any rating that results in an increased inspection frequency from the previous standard will be peer reviewed before conclusion by a GDP operations manager. MHRA does have a formal complaints process if sites wish to log an issue, however any concerns regarding the inspection process should be raised with the inspector.
Conditions of Holding a Wholesale Dealer’s Licence
The holder of a wholesale dealer’s licence must comply with certain conditions in relation to the wholesale distribution of medicinal products. These conditions are set out in regulations 43 – 45 of the Human Medicines Regulations 2012 [SI 2012/1916] (“the Regulations”). They require that the licence holder shall:
● comply with the guidelines on Good Distribution Practice (GDP);1
● ensure, within the limits of their responsibility as a distributor of medicinal products, the appropriate and continued supply of such medicinal products to pharmacies and persons who may lawfully sell such products by retail or who may lawfully supply them in circumstances corresponding to retail sale, so that the needs of patients in the UK are met;
● provide and maintain such staff, premises, equipment and facilities for the handling, storage and distribution of the medicinal products under the licence as are necessary to maintain the quality of, and ensure proper distribution of the medicinal products;
● inform the licensing authority of any proposed structural alteration to, or discontinued use of, premises to which the licence relates or premises which have been approved by the licensing authority;
● inform the licensing authority of any change to the Responsible Person;
● not sell or offer for sale or supply any medicinal product unless there is a marketing authorisation, Article 126a authorisation, certificate of registration or traditional herbal registration (“an authorisation”) for the time being in force in respect of that product; and the sale or offer for sale is in accordance with the provisions of that authorisation. This restriction on the holder of a wholesale dealer’s licence shall not apply to:
– the sale or offer for sale of a special medicinal product; and
– the export to an EEA State, or supply for the purposes of such export, of a medicinal product which may be placed on the market in that State without a marketing authorisation, Article 126a authorisation, certificate of registration or traditional herbal registration by virtue of legislation adopted by that State under Article 5(1) of the 2001 Directive; or
– the sale or supply, or offer for sale or supply, of an unauthorised medicinal product where the Secretary of State has temporarily authorised the distribution of the product under regulation 174 of the Regulations.
The holder of a wholesale dealer’s licence shall:
● keep such documents relating to the sale of medicinal products to which their licence relates as will facilitate the withdrawal or recall from sale of medicinal products in accordance with an emergency plan referred to below;
● have in place an emergency plan which will ensure effective implementation of the recall from the market of any relevant medicinal products where such recall is:
– ordered by the licensing authority or by the competent authority of any other EEA State, or
– carried out in co-operation with the manufacturer of, or the holder of the marketing authorisation for, the product in question;
● keep records in relation to the receipt, dispatch or brokering of medicinal products, of the date of receipt, the date of despatch, the date of brokering, the name of the medicinal product, the quantity of the product received, dispatched or brokered, the name and address of the person from whom the products were received or to whom they are dispatched, and the batch number of medicinal products bearing safety features referred to in point (o) of Article 54 of the 2001 Directive.2
Where the holder of a wholesale dealer’s licence imports from another EEA State for which they are not the holder of the marketing authorisation, Article 126a authorisation, certificate of registration or a traditional herbal registration of the product, then they shall notify the holder of that authorisation of their intention to import that product. In the case where the product is the subject of a marketing authorisation granted under Regulation (EC) No 726/2004, the holder of the wholesale dealer’s licence shall notify the EMA or for any other authorisation they shall notify the licensing authority. In both cases they will be required to pay a fee to the EMA in accordance with Article 76(4) of the 2001 Directive3 or the licensing authority as the case may be, in accordance with the Fees Regulations. These requirements will not apply in relation to the wholesale distribution of medicinal products to a person in a non-EEA country.
The licence holder, for the purposes of enabling the licensing authority to determine whether there are grounds for suspending, revoking or varying the licence, must permit a person authorised in writing by the licensing authority, on production of identification, to carry out any inspection, or to take any samples or copies, which an inspector could carry out or take under Part 16 (enforcement) of the Regulations.
The holder of a wholesale dealer’s licence must verify that any medicinal products they receive which are required by Article 54a of the Directive4 to bear safety features are not falsified. This does not apply in relation to the distribution of medicinal products received from a third country by a person for supply to a person in a third country. Any verification is carried out by checking the safety features on the outer packaging, in accordance with the requirements laid down in the delegated acts adopted under Article 54a(2) of the 2001 Directive.
The licence holder must maintain a quality system setting out responsibilities, processes and risk management measures in relation to their activities.
The licence holder must also immediately inform the licensing authority and, where applicable, the marketing authorisation holder, of medicinal products which the licence holder receives or is offered which the licence holder knows or suspects, or has reasonable grounds for knowing or suspecting, to be falsified.
Where the medicinal product is obtained through brokering, the licence holder must verify that the broker involved fulfils the requirements set out in the Regulations.
The licence holder must not obtain supplies of medicinal products from anyone except the holder of a manufacturer’s licence or wholesale dealer’s licence in relation to products of that description or the person who holds an authorisation granted by another EEA State authorising the manufacture of products of the description or their distribution by way of wholesale dealing. The supply must be in accordance with the principles and guidelines of good distribution practice. This does not apply in relation to the distribution of medicinal products directly received from a non-EEA country but not imported into the EU.
From 28th October 2013, where the medicinal product is directly received from a non-EEA country for export to a non-EEA country, the licensed wholesale dealer must check that the supplier of the medicinal product in the exporting non-EEA country is authorised or entitled to supply such medicinal products in accordance with the legal and administrative provisions in that country.
The holder of a wholesale dealer’s licence must verify that the wholesale dealer who supplies the product complies with the principles and guidelines of good distribution practices; or the manufacturer or importer who supplies the product holds a manufacturing authorisation.
The holder of a wholesale dealer’s licence may distribute medicinal products by way of wholesale dealing only to the holder of a wholesale dealer’s licence relating to those products, the holder of an authorisation granted by the competent authority of another EEA State authorising the supply of those products by way of wholesale dealing, a person who may lawfully sell those products by retail or may lawfully supply them in circumstances corresponding to retail sale; or a person who may lawfully administer those products. This does not apply in relation to medicinal products which are distributed by way of wholesale dealing to a person in a non-EEA country.
From 28th October 2013, where the medicinal product is supplied directly to persons in a non-EEA country the licensed wholesale dealer must check that the person that receives it is authorised or entitled to receive medicinal products for wholesale distribution or supply to the public in accordance with the applicable legal and administrative provisions of the non-EEA country concerned.
Where any medicinal product is supplied to any person who may lawfully sell those products by retail or who may lawfully supply them in circumstances corresponding to retail sale, the licence holder shall enclose with the product a document which makes it possible to ascertain:
● the date on which the supply took place;
● the name and pharmaceutical form of the product supplied;
● the quantity of product supplied;
● the names and addresses of the person or persons from whom the products were supplied to the licence holder; and
● the batch number of the medicinal products bearing the safety features referred to in point (o) of Article 54 of the 2001 Directive.
The holder of a wholesale dealer’s licence shall keep a record of the information supplied where any medicinal product is supplied to any person who may lawfully sell those products by retail or who may lawfully supply them in circumstance corresponding to retail sale for a minimum period of five years after the date on which it is supplied and ensure, during that period, that that record is available to the licensing authority for inspection.
The wholesale dealer’s licence holder shall at all times have at their disposal the services of a responsible person who, in the opinion of the licensing authority has knowledge of the activities to be carried out and of the procedures to be performed under the licence which is adequate for performing the functions of responsible person; and has experience in those procedures and activities which is adequate for those purposes.
The functions of the responsible person shall be to ensure, in relation to medicinal products, that the conditions under which the licence has been granted have been, and are being, complied with and the quality of medicinal products which are being handled by the wholesale dealer’s licence holder are being maintained in accordance with the requirements of the marketing authorisations, Article 126a authorisations, certificates of registration or traditional herbal registrations applicable to those products.
The standard provisions for wholesale dealer’s licences, that is, those provisions which may be included in all licences unless the licence specifically provides otherwise, insofar as those licences relate to relevant medicinal products, shall be those provisions set out in Part 4 of Schedule 4 of the Regulations.
The licence holder shall not use any premises for the purpose of the handling, storage or distribution of relevant medicinal products other than those specified in their licence or notified to the licensing authority by them and approved by the licensing authority.
The licence holder shall provide such information as may be requested by the licensing authority concerning the type and quantity of any relevant medicinal products which they handle, store or distribute.
Where and insofar as the licence relates to special medicinal products to which regulation 167 of the Regulations apply which do not have a UK or EMA authorisation and are commonly known as “specials” (refer to Guidance Note 14), the licence holder shall only import such products from another EEA State in response to an order which satisfies the requirements of regulation 167 of the Regulations; and where the following conditions are complied with:
● No later than 28 days prior to each importation of a special medicinal product, the licence holder shall give written notice to the licensing authority stating their intention to import that special medicinal product and stating the following particulars:
– the name of the medicinal product, being the brand name or the common name, or the scientific name, and any name, if different, under which the medicinal product is to be sold or supplied in the United Kingdom;
– any trademark or name of the manufacturer of the medicinal product,
– in respect of each active constituent of the medicinal product, any international non-proprietary name or the British approved name or the monograph name or, where that constituent does not have an international non-proprietary name, a British approved name or a monograph name, the accepted scientific name or any other name descriptive of the true nature of that constituent,
– the quantity of medicinal product which is to be imported which shall not exceed more, on any one occasion, than such amount as is sufficient for 25 single administrations, or for 25 courses of treatment where the amount imported is sufficient for a maximum of three months’ treatment, and
– the name and address of the manufacturer or assembler of that medicinal product in the form in which it is to be imported and, if the person who will supply that medicinal product for importation is not the manufacturer or assembler, the name and address of such supplier.
● Subject to the next bullet point below, the licence holder shall not import the special medicinal product if, before the end of 28 days from the date on which the licensing authority sends or gives the licence holder an acknowledgement in writing by the licensing authority that they have received the notice referred to in the bullet point above, the licensing authority have notified them in writing that the product should not be imported.
● The licence holder may import the special medicinal product referred to in the notice where they have been notified in writing by the licensing authority, before the end of the 28 day period referred to in the bullet point above, that the special medicinal product may be imported.
● Where the licence holder sells or supplies special medicinal products, they shall, in addition to any other records which they are required to make by the provisions of their licence, make and maintain written records relating to the batch number of the batch of the product from which the sale or supply was made and details of any adverse reaction to the product so sold or supplied of which they become aware.
● The licence holder shall import no more on any one occasion than such amount as is sufficient for 25 single administrations, or for 25 courses of treatment where the amount imported is sufficient for a maximum of three months’ treatment, and on any such occasion shall not import more than the quantity notified to the licensing authority in the notification of intention to import.
● The licence holder shall inform the licensing authority forthwith of any matter coming to their attention which might reasonably cause the licensing authority to believe that the medicinal product can no longer be regarded either as a product which can safely be administered to human beings or as a product which is of satisfactory quality for such administration.
● The licence holder shall not issue any advertisement, catalogue or circular relating to the special medicinal product or make any representations in respect of that product.
● The licence holder shall cease importing or supplying a special medicinal product if they have received a notice in writing from the licensing authority directing that, as from a date specified in that notice, a particular product or class of products shall no longer be imported or supplied.
The licence holder shall take all reasonable precautions and exercise all due diligence to ensure that any information they provide to the licensing authority which is relevant to an evaluation of the safety, quality or efficacy of any medicinal product for human use which they handle, store or distribute is not false or misleading in a material particular.
Where a wholesale dealer’s licence relates to exempt advanced therapy medicinal products the licence holder shall keep the data for the system for the traceability of the advanced therapy medicinal products for such period, being a period of longer than 30 years, as may be specified by the licensing authority.
The Standard Provisions also require the holder of a wholesale dealer’s licence that relates to exempt advanced therapy medicinal products to obtain supplies of exempt advanced therapy medicinal products only from the holder of a manufacturer’s licence in respect of those products or the holder of a wholesale dealer’s licence in respect of those products.
The licence holder must:
● distribute an exempt advanced therapy medicinal product by way of wholesale dealing only to the holder of a wholesale dealer’s licence in respect of those products; or a person who may lawfully administer those products, and solicited the product for an individual patient;
● establish and maintain a system ensuring that the exempt advanced therapy medicinal product and its starting and raw materials, including all substances coming into contact with the cells or tissues it may contain, can be traced through the sourcing, manufacturing, packaging, storage, transport and delivery to the establishment where the product is used;
● inform the licensing authority of any adverse reaction to any exempt advanced therapy medicinal product supplied by the holder of the wholesale dealer’s licence of which the holder is aware;
● keep the data for ensuring traceability for a minimum of 30 years after the expiry date of the exempt advanced therapy medicinal product or longer as specified by the licensing authority;
● ensure that the data for ensuring traceability will, in the event that the licence is suspended, revoked or withdrawn or the licence holder becomes bankrupt or insolvent, be held available to the licensing authority by the holder of a wholesale dealer’s licence for the same period that the data has to be kept; and
● not import or export any exempt advanced therapy medicinal product.
Appointment and Duties of the Responsible Person
Title VII of the Directive on the Community code relating to medicinal products for human use (Directive 2001/83/EC) obliges holders of a distribution authorisation to have a “qualified person designated as responsible”. Regulation 45 of the Human Medicines Regulations 2012 [SI 2012/1916] state the requirement for a Responsible Person (RP) within the UK.
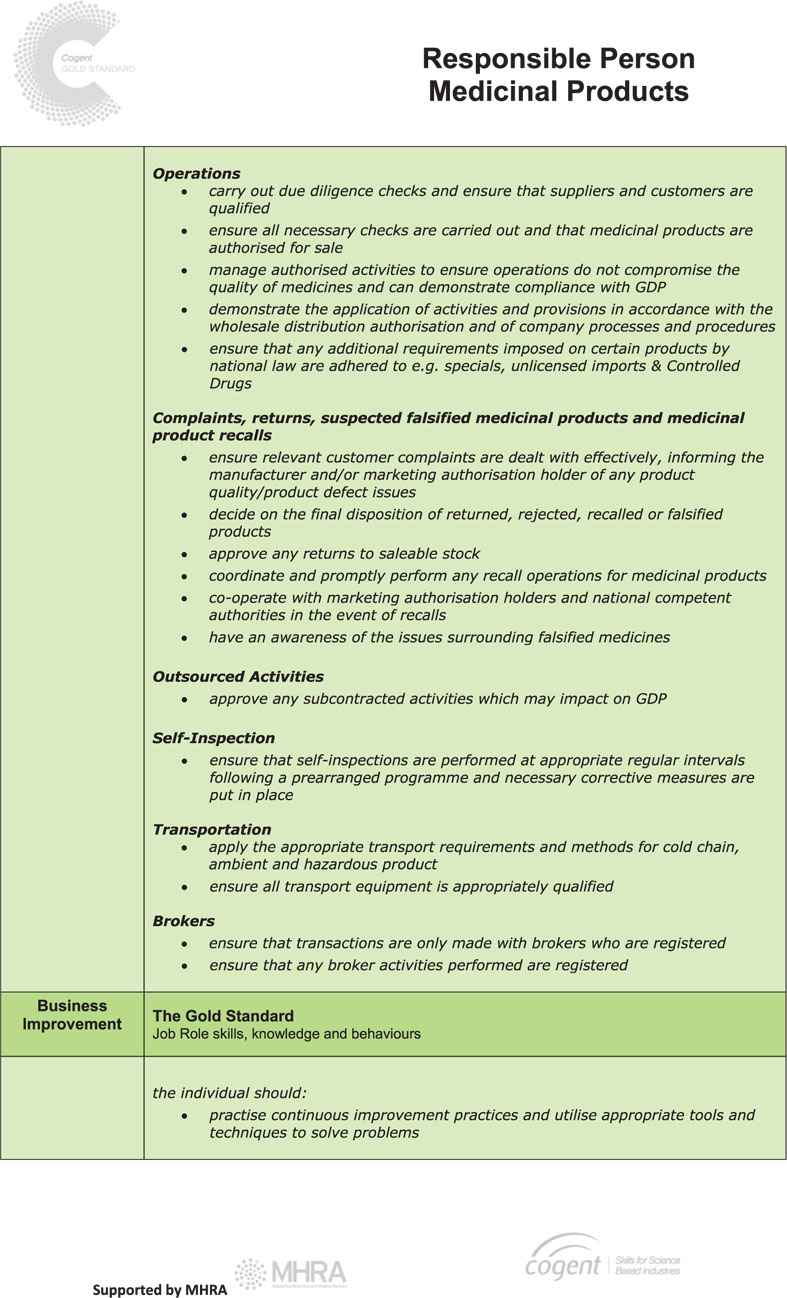
The RP is responsible for safeguarding product users against potential hazards arising from poor distribution practices as a result, for example, of purchasing suspect products, poor storage or failure to establish the bona fides of purchasers. The duties of a RP include:
● to ensure that the provisions of the licence are observed
● to ensure that the guidelines on Good Distribution Practice (GDP) are complied with
● to ensure that the operations do not compromise the quality of medicines
● to ensure that an adequate quality system is established and maintained
● to oversee audit of the quality system and to carry out independent audits
● to ensure that adequate records are maintained
● to ensure that all personnel are trained
● to ensure full and prompt cooperation with marketing authorisation holders in the event of recalls.
In order to carry out his duties, the RP should be resident in the UK and have a clear reporting line to the licence holder or MD. The RP should have personal knowledge of the products traded under the licence and the conditions necessary for their safe storage and distribution. The RP should have access to all areas, sites, stores and records which relate to the licensable activities and regularly review and monitor all such areas, etc. and the standards achieved.
If the RP is not adequately carrying out those duties, the licensing authority may consider the suspension of the licence, withdrawal of acceptance of the RP on that licence and the acceptability on any other licence.
The RP does not have to be an employee of the licence holder but must be available to the licence holder when required. Where the RP is not an employee, there should be a written contract specifying responsibilities, duties, authority and so on.
In the case of small companies, the licensing authority may accept the licence holder as the nominated RP. In larger companies, however, this is not desirable.
There is no statutory requirement for the RP to be a pharmacist.
The RP should have access to pharmaceutical knowledge and advice when it is required, and have personal knowledge of:
● The relevant provisions of the Human Medicines Regulations 2012 [SI 2012/1916].
● Directive 2001/83/EC as amended on the wholesale distribution of medicinal products for human use.
● The EU Guidelines on Good Distribution Practice of Medicinal Products for Human Use (2013/C 343/01).
● The conditions of the wholesale dealer’s licence for which nominated.
● The products traded under the licence and the conditions necessary for their safe storage and distribution.
● The categories of persons to whom products may be distributed.
Where the RP is not a pharmacist or eligible to act as a Qualified Person (QP) (as defined in Directive 2001/83/EC as amended), the RP should have at least one year’s practical experience in both or either of the following areas:
● Handling, storage and distribution of medicinal products.
● Transactions in or selling or procuring medicinal products. In addition, the RP should have at least one year’s managerial experience in controlling and directing the wholesale distribution of medicinal products on a scale, and of a kind, appropriate to the licence for which nominated.
To carry out responsibilities, the RP should:
● Have a clear reporting line to either the licence holder or the Managing Director.
● Have access to all areas, sites, stores, staff and records relating to the licensable activities being carried out.
● Demonstrate regular review and monitoring of all such areas, sites and staff etc. or have delegated arrangements whereby the RP receives written reports that such delegated actions have been carried out on behalf of the RP in compliance with standard operating procedures and GDP.
Where arrangements are delegated, the RP remains responsible and should personally carry out the delegated functions at least once a year.
● Focus on the management of licensable activities, the accuracy and quality of records, compliance with standard operating procedures and GDP, the quality of handling and storage equipment and facilities, and the standards achieved.
● Keep appropriate records relating to the discharge of the RP responsibilities.
Where the licence covers a number of sites, the RP may have a nominated deputy with appropriate reporting and delegating arrangements. However, the RP should be able to demonstrate to the licensing authority that the necessary controls and checks are in place.
The licence holder should ensure that there is a written standard operating procedure for receiving advice and comment from the RP and recording the consequent action taken as may be necessary.
Should it prove impossible to resolve a disagreement between the licence holder and the RP, the licensing authority should be consulted.
Whilst a joint referral is clearly to be preferred, either party may approach the licensing authority independently. If an RP finds difficulty over performing statutory responsibilities or the activities being carried out under the licence, the licensing authority should be consulted in strict confidence.
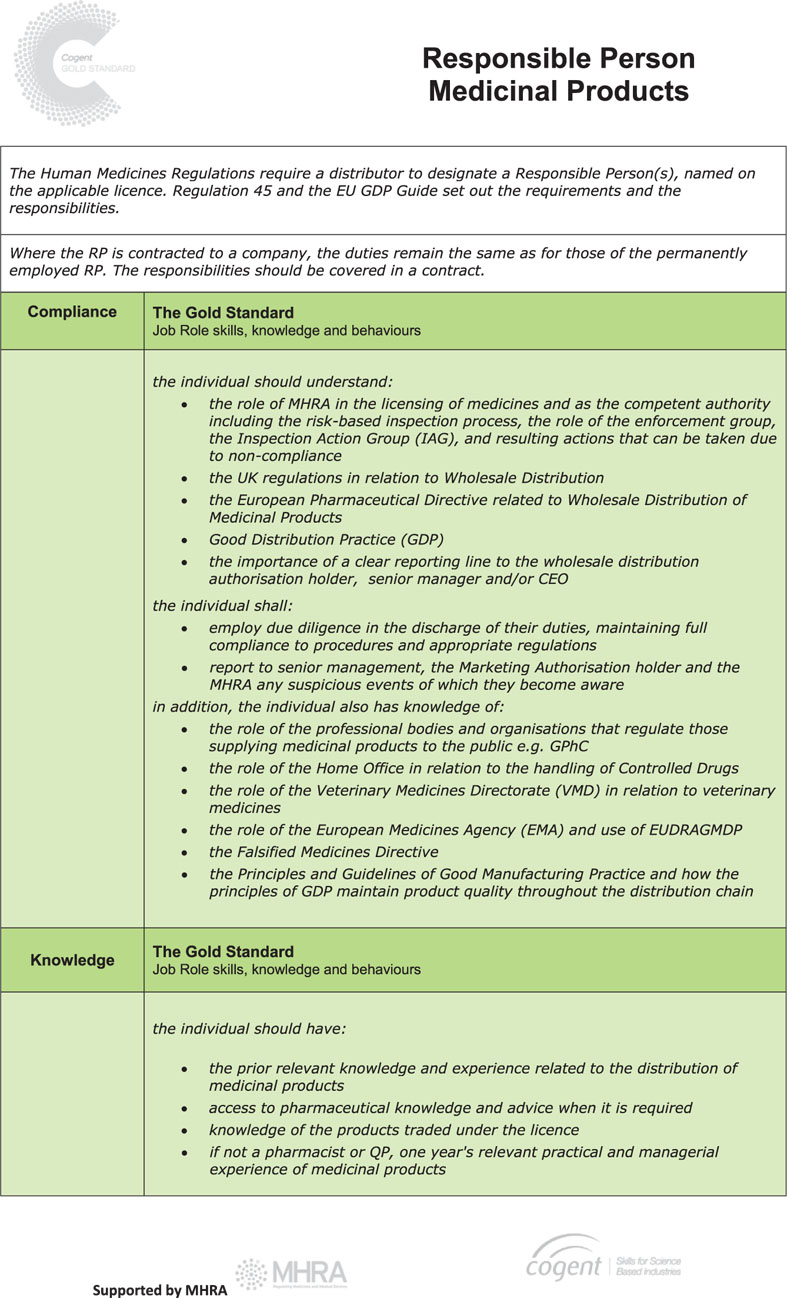
The Responsible Person Gold Standard
The Human Medicines Regulations 2012 require holders of a wholesale dealer’s licence to designate and ensure that there is available at all times at least one person, referred to in the regulations as the “responsible person”, who in the opinion of the licensing authority:
(a) has knowledge of the activities to be carried out and of the procedures to be performed under the licence; and
(b) has adequate experience relating to those activities and procedures.
Guidance on the role and responsibilities of the Responsible Person (RP) is set out in Chapter 2 of the EU Good Distribution Practice Guide and these remain the same irrespective of whether the RP is a permanent or contracted employee of a company.
The RP plays a vital part in ensuring the quality and the integrity of medicinal products are maintained throughout the distribution chain and it is essential that they have the right knowledge, demonstrate competence and deploy the right skills so that patients and healthcare professionals have the confidence and trust to use medicines.
In order to facilitate this and to standardise the requirements for individuals operating as, or aspiring to be, a RP, Cogent (the national skills body for the science industries) has, following extensive discussion with pharmaceutical companies and MHRA, published a new Gold Standard role profile for the RP1.
This sets out an industry-agreed framework that identifies the skills required in four competency areas and includes not only traditional qualifications and technical requirements but also the behavioural skills necessary to do the job to a high standard.


UK Guidance on EU Guide Chapter 1 – Quality Management
Quality Management
A consistent focus on quality is of prime importance for all wholesale distributors in order to maintain an effective and efficient business that meets customer needs and ensures product quality is maintained.
Quality management is as much a mind-set as an activity in itself, with the aim of achieving quality processes that permeate throughout all distribution activities, with the ultimate goal of ensuring patient safety.
Quality System
The quality system is the vehicle by which quality management is delivered and it should be all encompassing. However, this doesn’t mean that it has to be complicated with its size and complexity being proportionate to the distribution activities being undertaken.
Chapter 1 of the EC Guide on Good Distribution Practice (GDP) gives a detailed breakdown of the areas that should be covered.
Quality Risk Management (QRM)
QRM is not new to wholesale distributors, as informal systems have always been in place as a matter of routine. However GDP now require that the process be documented.
QRM is essentially the identification and control of risks to product quality through the evaluation of the activities that are being performed.
Underpinning this is that the evaluation of the risk is based on knowledge and experience of the process and ultimately links to the protection of the patient.
The level of effort, formality and documentation of the process should be commensurate with the level of risk.
GDP refers to ICH Q9 as a useful guidance document on QRM. This includes the principles and concepts of QRM with Annex 1 & 2 identifying various tools and techniques; www.ich.org
Change control
Change control is a formal process whereby changes to a process are identified, planned and introduced in a controlled way.
A typical change control process may consist of several stages;
● Submission of a documented change control request to management.
● Consideration and evaluation of the request by management. This evaluation should consider if the change poses any risks to public health or if there are any impacts on other processes and procedures. Actions identified at this stage should be resolved prior to approval.
● Implementation of the change.
This is a live process and unforeseen events during implementation should be subject to a further assessment of risk,
● Review of Effectiveness. After an appropriate period an objective review of the effectiveness of the change should take place. This should consider both successes as well as any problems that have arisen, with any lessons learnt being used to improve the process for future changes.
Deviation management and Corrective and Preventative Actions (CAPA)
Distributors should prepare for circumstances when things go wrong.
When this occurs the identification of control measures should be a priority, particularly where an unplanned event may have (or has had) a significant impact on public health.
Depending on the severity of the event, there may be a process of ongoing control, requiring initial corrective actions to deal with the immediate fallout from the event through to the identification of the root cause of the problem and proactive preventative actions to avoid reoccurrence.
During investigations it is very important that the root cause of the problem is identified, as what may initially be deemed as for example, “operator error” may actually be attributable to system weaknesses elsewhere.
Corrective and Preventative actions (CAPA), are an integral part of the change control and deviation management processes; reducing the likelihood or severity of a risk event and creating an opportunity to improve and reduce the likelihood of the deviation occurring again.
Corrective actions are identified after a deviation with preventative actions being identified following an event and before a change is implemented.
Management review and monitoring
To ensure the effectiveness and continual improvement of the quality management system, there should be a process in place for regular management review.
This review should actively involve company management and key personnel such as the Responsible Person.
In very small organisations the process of review may require taking a step back and objectively reviewing the operational activities and supporting procedures.
Questions to be considered during the management review may include:
● Is the quality system effective, does it reflect the current business model?
● Are activities being carried out in accordance with the principles of Quality Management, or are there opportunities for improvement with some processes requiring modification?
● Has the regulatory landscape changed? Has legislation been amended or guidance published which means that activities need to be reviewed?
● Is the business operating effectively, are the right products getting to the right customers at the right time? What improvements can be made to address customer complaints?
● If third parties are contracted to provide services, are they complying with their contractual obligations?
● Have there been any unforeseen problems or events and has effective CAPA been implemented?
● Have planned changes been effective?
● Are there staff training or staff re-training issues?
The outcome of each management review should be documented, result in a CAPA plan where necessary and should be effectively communicated to staff.
Controls on Certain Medicinal Products
Products containing Ephedrine and Pseudoephedrine
The Drug Licensing and Compliance Unit at the Home Office is informing exporters of pseudoephedrine and ephedrine (both for human or veterinary use) outside of the European Union of amendments to the existing Drug Precursor Chemical Regulations which came into effect on the 30 December 2013. A summary of the key changes can be found on GOV.UK1.
Drugs useable in execution by lethal injection
In December 2011, the European Union adopted a new EU-wide control on the export of certain drugs usable in execution by lethal injection.
These controls which came into force on 21 December 2011 were adopted as an amendment to Annex III of Council Regulation (EC) 1236/2005 concerning trade in certain goods which could be used for capital punishment, torture or other cruel, inhuman or degrading treatment or punishment.
You can download the full text of Council Regulation (EU) No 1352/2011 from the Eudralex website2.
As a result of these EU imposed controls, exporters need to seek appropriate permission from national export control authorities to export to any destination outside the EU, short and intermediate acting barbiturate anaesthetic agents including, but not limited to, the following:
● amobarbital (CAS RN 57-43-2)
● amobarbital sodium salt (CAS RN 64-43-7)
● pentobarbital (CAS RN 76-74-4)
● pentobarbital sodium salt (CAS 57-33-0)
● secobarbital (CAS RN 76-73-3)
● secobarbital sodium salt (CAS RN 309-43-3)
● thiopental (CAS RN 76-75-5)
● thiopental sodium salt (CAS RN 71-73-8), also known as thiopentone sodium
The control also applies to products containing one or more of the above. These controls are intended to apply to finished products – in other words, those that are packaged for human or veterinary use. It is not intended that they should apply to raw materials or to intermediate products (ie products that require further processing to make them suitable for human or veterinary use).
The relevant national export licensing authority responsible for administering these controls in the UK is the Export Control Organisation (ECO), part of the Department for Business, Innovation and Skills (BIS). The ECO is responsible for licensing items that are judged to be of a ‘strategic’ nature, including goods used in torture.
Further information can be found on the GOV.UK web site3.
Control of Lisdexamfetamine, Tramadol, Zaleplon, Zopiclone and Reclassification of Ketamine
On 10 June 2014, the Parliamentary Order controlling, and in the case of ketamine reclassifying the following drugs, came into force.
● lisdexamfetamine
● tramadol
● zaleplon
● zopiclone
● medicines containing these substances.
The Order, available on legislation.gov.uk (external link), controls:
● lisdexamphetamine as a Class B drug
● tramadol as a Class C drug
● zopiclone and zaleplon as Class C drugs.
The Order also reclassifies ketamine as a Class B drug under the Misuse of Drugs Act 1971.
Companies who possess, supply or produce lisdexamfetamine, tramadol, zaleplon, zopiclone (or medicines containing these substances) need to get the correct licences from the Home Office.
More information on how to apply for a licence, and how much they cost are available on www.GOV.UK at https://www.gov.uk/controlled-drugs-licences-fees-and-returns or by calling the Duty Compliance Officer on 020 7035 8972.
Companies without the correct licences are at risk of prosecution.
The listed drugs will be scheduled, alongside their control, as follows to ensure that they remain available for use in healthcare:
● lisdexamfetamine (a drug which converts to dexamfetamine when administered orally and used as second line treatment for ADHD) will be listed in Schedule 2 alongside dexamfetamine
● tramadol will be listed in Schedule 3 but exempted from the safe custody requirements. Full prescription writing requirements under regulation 15 will apply to its use in healthcare,
● zopiclone and zaleplon will be listed in Part 1 of Schedule 4 alongside zolpidem.
Ketamine is not being rescheduled immediately. In line with the Advisory Council on the Misuse of Drugs’ (ACMD) advice, the Home Office will carry out a public consultation later this year to assess the impact of rescheduling ketamine to Schedule 2.
A final decision on the appropriate schedule for ketamine will be made after the consultation. Until then ketamine will remain a Schedule 4 Part 1 drug.
Home Office Circular 008/2014: A change to the Misuse of Drugs Act 1971 – Control of NBOMes, Benzofurans, Lisdexamphetamine, Tramadol, Zopiclone, Zaleplon and Reclassification of Ketamine1.
Best Practice Temperature Monitoring
Why control and monitor temperature?
1 Manufacturers subject their products to stability studies that are used to determine appropriate storage conditions including those for temperature. These conditions are therefore specific for each product, and wholesalers should refer to manufacturers’ information when deciding the storage conditions to use.
Medicinal products experiencing an adverse temperature may undergo physical, chemical or microbiological degradation. In the most serious of cases this may lead to conversion of the medicine to ineffective or harmful forms. The ability to detect these changes may not appear until the medicine is consumed, and it is therefore essential that appropriate temperature conditions are controlled and monitored throughout each step of the supply chain.
Control and monitoring of storage areas
2 Where medicines are stored that may be required in an emergency then contingency measures should be put in place such as linking essential equipment in a large warehouse to a source of emergency power. These emergency measures should be routinely tested, such as the confirmation of restoration of stored data and settings when emergency power supply is activated and after normal power is resumed. For these products there should be a system in place to ensure that on-call personnel are notified in the event of power failure or temperature alarms being triggered including notification outside of normal working hours.
3 The application of Mean Kinetic Temperature (MKT) to temperature monitoring of wholesale products is only appropriate where an acceptable MKT value is provided by the MA holder for a specific product, and the recording of temperature can be confirmed to be consistent and complete from the moment of leaving the manufacturer’s premises. In practice the application of MKT fails where a complete chain of temperature recording cannot be allocated to a specific consignment of a product. Attempts to apply MKT have been proposed by wholesalers as an alternative to having adequate temperature control within their warehouses as well as attempting to downgrade the impact of temperature excursions. The use of MKT in the wholesale environment without robust supporting information and methodology is therefore discouraged.
SMALL REFRIGERATORS
4 Refrigerators used to store pharmaceuticals should be demonstrated to be fit for purpose. In the simplest of cases a new off-the–shelf refrigerator installed according to the manufacturer’s instructions and temperature monitored with an appropriate device may be considered appropriately qualified for storing cold chain product that is shown to be unaffected by minor temperature excursions. A refrigerator used for holding more susceptible stock such as biological products will require more extensive qualification.
5 In addition to temperature mapping and monitoring there should be safeguards to preserve appropriate storage conditions. Some small refrigerators are purported to be medical or pharmaceutical refrigerators but this on its own does not automatically render them suitable for wholesale use. The refrigerator should be capable of restoring the temperature quickly after the door has been opened and without danger of overshooting to extreme cold. This could be assisted by an internal fan and good shelf design which enables an efficient air flow. There should be no internal ice box and no internal temperature dials capable of being inadvertently knocked and adjusted.
6 Storage practices for using small refrigerators should include consideration of segregation of stock with different status, e.g. incoming, quarantine, returned and outgoing stock. Sufficient space should be maintained to permit adequate air circulation and product should not be stored in contact with the walls or on the floor of the refrigerator. If the refrigerator is filled to capacity the effect on temperature distribution should be investigated. Where non-refrigerated items are introduced to the refrigerator, such as non-conditioned gel packs, the impact of introducing these items should be assessed regarding the increase in temperature they cause.
LARGE COMMERCIAL REFRIGERATORS AND WALK-IN COLD ROOMS
7 These should be of appropriate design, suitably sited and be constructed with appropriate materials. The design should ensure general principles of GDP can be maintained, such as segregation of stock. Condensate from chillers should not be collected inside the unit and there should be a capability to carry out routine maintenance and service activities as much as possible from outside the unit. The temperature should be monitored with an electronic temperature-recording device that measures load temperature in one or more locations depending on the size of the unit, and alarms should be fitted to indicate power outages and temperature excursions.
FREEZERS
8 The same general principles apply to freezers as apply to other cold chain storage units above. Walk-in freezers pose a significant operator health and safety risk, and the impact of ways of working should be reviewed with consideration of risk to causing temperature excursions.
Calibration of temperature monitoring devices
9 In order to have confidence in temperature readings monitoring devices should be calibrated to demonstrate they have appropriate accuracy and precision. Temperate storage thermometers should be capable of reading ±1°C, and cold chain devices capable of reading ±0.5°C. Calibration should extend across the whole of the working range, so for a temperate storage range of 15°C to 25°C the calibration range may be 10°C to 30°C to allow the thermometer to be used in assessing temperature excursions or to be used in temperature mapping exercises. Results of the calibration exercise should be presented in a report or calibration certificate approved by the calibrator and demonstrated to be appropriate for use by the wholesaler. The certificate should include the following details:
● Serial number of the calibrated instrument
● Serial numbers of test instruments
● Traceability to national or international calibration standards
● Calibration test method used
● ISO or equivalent registration details of calibration laboratory
● Date of calibration
● Calibration results
● Unique certificate number
● Approval of results by calibrator.
Where a temperature monitoring device reads temperature from a main monitoring unit plus a remote probe it should be clear from the calibration certificate which part of the device the calibration refers to. Calibration should be carried out annually, and where adjustments are made to the equipment as part of calibration an assessment of accuracy and precision should be made prior to adjustment in addition to following adjustment. On completion a suitable representative from the wholesaler should approve the calibration indicating its suitability for use.
Under the legislation on advertising medicines, companies may only provide free samples to persons qualified to prescribe the medicine. Samples may only be supplied in response to a signed and dated request from the prescriber and must be appropriately labelled and accompanied by a copy of the Summary of Product Characteristics (SPC). The company must have adequate procedures for control and accountability for all samples. See section 6.12 of MHRA’s Blue Guide for details of the legal requirements.
MHRA is aware that in some cases sales representatives receive samples to fill prescriber requests and that the medicinal products are delivered to them by colleagues or by couriers. Either way, the storage and delivery arrangements for these medicinal products must be validated to ensure the medicinal product will be transported expeditiously under controlled Good Distribution Practice (GDP) conditions and in accordance with labelled storage requirements at all times. It is highly unlikely that samples requiring refrigeration will meet these requirements. With regards to storage it is not acceptable for samples to be stored in the representative’s home (on unlicensed premises, which are not GDP compliant), lacking appropriate storage facilities, security and controls to maintain the quality of the medicines and provide an audit trail.
Likewise distribution of samples involving delivery in a representative’s vehicle that has no provision for maintaining correct storage conditions is also unacceptable. Temperatures in a car boot in high summer could reach 50 degrees Celsius or go below 0 degrees Celsius in winter. The practice of providing sales representatives with samples of medicinal products which they retain for onward distribution is therefore unlikely to be acceptable due to the storage and transport difficulties outlined above. The only reason for which sales representatives may hold samples on a long-term basis is for the purpose of product identification. In this regard procedures must be in place to ensure accountability for any such stock and to ensure no packs are provided to healthcare professionals.
Handling Returns of Non-defective Medicinal Products
Any person acting as a wholesale distributor must hold a wholesale dealer’s licence (WDA(H)). Article 80(g) of Directive 2001/83/EC provides that distributors of human medicines must comply with the principles of and guidelines for good distribution practice (GDP).
The Commission has revised its guidelines for GDP which are now contained in the Guidelines on Good Distribution Practice of Medicinal Products for Human Use (2013/C 343/01). Paragraph 6.3 of GDP refers to returned medicinal products, the key elements being that; “products that have left the premises of the distributor should only be returned to saleable stock if…the medicinal products are in their unopened and undamaged secondary packaging and are in good condition; have not expired and have not been recalled; it has been demonstrated by the customer that the medicinal products have been transported, stored and handled in compliance with their specific storage requirements; and they have been examined and assessed by a sufficiently trained and competent person authorised to do so;”
MHRA re-affirm that a licensed site can only be interpreted as being under full GDP control at a licensed wholesale dealer’s site. This applies to all categories of medicines. Medicinal products held in unlicensed storage and distribution sites are not considered to be within the licensed wholesale distribution network.
Returns from a licensed wholesale dealer’s site
MHRA will adopt a pragmatic approach to the return of non defective medicinal products for those products returned from a customer operating from a licensed wholesale dealer’s site.
In such circumstances, the return should be completed as expeditiously as possible and the most expedient and appropriate method of transportation must be used.
Returns from an unlicensed site
AMBIENT
For those non defective ambient medicinal products returned from an unlicensed site, the return should be completed within five days, including transport.
REFRIGERATED
For those non defective refrigerated medicinal products returned from an unlicensed site, the return should be completed within 24 hours, including transport.
The Responsible Person or the authorised person must be able to demonstrate evidence of “full knowledge” of the storage whilst at the unlicensed site, including transportation to and from the site.
The EU GDP Guidelines define wholesale distribution as;
“…all activities consisting of procuring, holding, supplying or exporting medicinal products…” The annexed Glossary of Terms defines holding as “storing medicinal products”.
Medicinal products should therefore only be stored on premises that are covered by a wholesale dealer’s licence.
However, there are certain cases where medicinal products are held for short periods of time during transportation and prior to onward shipment e.g. in the transportation vehicle at motorway service stations or in overnight freight depots.
In such instances it has been determined that, as a matter of policy, a site does not have to be named on a licence where ambient products are stored for less than 36 hours.
Sites holding ambient products in excess of 36 hours must be licensed
This policy applies only where ownership of the products has not been transferred to the person carrying out the storage activities.
Where ownership has been transferred, this is supply and as such the receiving site must be licensed.
It is also important to note that, where wholesaling activities other than storage are being carried out, the site should be named on the relevant licence. This includes the handling of returned goods and where decisions are made regarding suitability for resale, as well as the usual activities of picking against orders.
Sites where refrigerated products are held, even when this is for less than 36 hours, must be licensed
The exception will be where these products are transported and stored overnight in continuously refrigerated vehicles.
The provisions of Chapter 9.2 of the EU GDP Guidelines must also be observed.
Qualification of Customers and Suppliers
Before commencing wholesale dealing activities with a customer or supplier (trading partners), wholesale dealers should ensure that their proposed trading partners are entitled to trade with them. Checks should demonstrate that trading partners either hold the required manufacturing and wholesale dealer’s licence where necessary or that they are entitled to receive medicines for the purpose of retail supply or for use in the course of their business.
Wholesale dealers should request those trading partners that wholesale or manufacture human medicines to supply a copy of their licence before trading commences.
Subsequent and continuing bona fides checks can be made by checking against the Register of licence holders on MHRA’s website.
Qualification of suppliers
Maintaining the integrity of the supply chain is one of the most important aspects of wholesale distribution. A robust fully documented system to ensure medicines are sourced appropriately must be in place and subjected to regular review. Wholesalers must ensure their suppliers are appropriately licenced to supply medicines. The qualification of suppliers requires the following steps to be fully complaint.
● The licence of the supplier should be viewed, either a copy obtained from the company or the details can be viewed on the MHRA’s website that has registers of wholesalers and manufacturers.1 Whilst MHRA registers are updated regularly they must not be relied on as a sole means of qualifying suppliers’ authority to supply.
● The EudraGMDP website has daily updates from the MHRA and contains all current live licences. The EudraGMDP website does not however contain all the information on a WDA(H) that companies need to fully qualify suppliers and must not be relied on as a sole means of qualifying suppliers’ authority to supply.2
● For supplies from other EEA member states the same checks should be made on EudraGMDP and via licences that have been translated. The translated licences should be translated and authenticated as such by a notary.
Compliance with GDP
Wholesalers must verify that wholesale suppliers comply with the principles and guidelines of good distribution practices.
● The GDP certificate of the wholesaler should be viewed on the EudraGMDP website. The date of the certificate expiry should be noted.
● If there is no GDP certificate available then other evidence of GDP compliance by the wholesale supplier should be obtained, such as a copy of the inspection close out letter confirming GDP compliance.
Routine re-qualification
Wholesalers must be aware of issues that could affect their suppliers’ continued authority to supply. The following should be carried out:
● Regular checks at least twice a month of MHRA’s list of suspended licence holders.
● Regular checks on EudraGMDP website for issued GMP and GDP statements of non-compliance.
● At least annually, a documented full re-qualification of suppliers.
Due diligence
When entering into a new contract with new suppliers, the wholesale distributor should carry out ‘due diligence’ checks in order to assess the suitability, competence and reliability of the other party. This could include checking the financial status of the supplier.
A “falsified medicinal product” means any medicinal product with a false representation of:
(a) its identity, including its packaging and labelling, its name or its composition (other than any unintentional quality defect) as regards any of its ingredients including excipients and the strength of those ingredients;
(b) its source, including its manufacturer, its country of manufacturing, its country of origin or its marketing authorisation holder; or
(c) its history, including the records and documents relating to the distribution channels used.
The supply of falsified medicines is a global phenomenon and one which MHRA takes very seriously. Falsified medicines represent a threat to the legitimate UK supply chain and to patient safety. They are fraudulent and may be deliberately misrepresented with respect to identity, composition and/or source. Falsification can apply to both innovator and generic products, prescription and self-medication, as well as to traditional herbal remedies. Falsified medicines may include products with the correct ingredients but fake packaging, with the wrong ingredients, without active ingredients or with insufficient active ingredients, and may even contain harmful or poisonous substances.
The supply and distribution of medicines is tightly controlled within the European Community.
All licensed wholesalers must comply with the Community’s agreed standards of good distribution practice (GDP) and there exist strict licensing and regulatory requirements in UK domestic legislation to safeguard patients against potential hazards arising from poor distribution practices: for example, purchasing suspect or falsified products, failing to establish the “bona fides” of suppliers and purchasers, inadequate record keeping, and so on.
Section 6.4 of the EU Guide to GDP is of principal importance to wholesale dealers. This states:
“Wholesale distributors must immediately inform the competent authority and the marketing authorisation holder of any medicinal products they identify as falsified or suspect to be falsified1. A procedure should be in place to this effect. It should be recorded with all the original details and investigated.
Any falsified medicinal products found in the supply chain should immediately be physically segregated and stored in a dedicated area away from all other medicinal products. All relevant activities in relation to such products should be documented and records retained.”
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


