UK Guidance on the Manufacture, Importation and Distribution of Active Substances
EU Directive 2001/83/EC lays down the rules for the manufacture, import, marketing and supply of medicinal products and ensures the functioning of the internal market for medicinal products while safeguarding a high level of protection of public health in the EU.
The falsification of medicinal products is a global problem, requiring effective and enhanced international coordination and cooperation in order to ensure that anti-falsification strategies are more effective, in particular as regards sale of such products via the Internet. To that end, the Commission and Member States are cooperating closely and supporting ongoing work in international fora on this subject, such as the Council of Europe, Europol and the United Nations. In addition, the Commission, working closely with Member States, is cooperating with the competent authorities of third countries with a view to effectively combating the trade in falsified medicinal products at a global level.
Active substances are those substances which give a medicinal product its therapeutic effect. They are the Active Pharmaceutical Ingredient (API).
Falsified active substances and active substances that do not comply with applicable requirements of Directive 2001/83/EC pose serious risks to public health.
The Falsified Medicines Directive 2011/62/EU amends Directive 2001/83/EC in order to facilitate the enforcement of and control of compliance with Union rules relating to active substances. It makes a number of significant changes to the controls on active substances intended for use in the manufacture of a medicinal product for human use.
A number of new terms have been introduced into the 2001 Directive by the Falsified Medicines Directive, including “falsified medicinal product” and “active substance”. The aim of this is to ensure that other amendments introduced by the Falsified Medicines Directive are consistently interpreted and applied across the EU.
The 2001 Directive has been amended to permit the European Commission to adopt the following:
● the principles and guidelines of good manufacturing practice for active substances, by means of a delegated act; and
● the principles of good distribution practice for active substances, by means of adopted guidelines.
To provide a greater level of control, and transparency of supply, for active substances within the European Community manufacturers, importers and distributors of active substances have to notify the relevant competent authorities of their activities and provide certain details. In the UK this will be MHRA. The competent authority has an obligation to enter these details into a Community Database (EudraGMDP) following the determination of a successful application for registration. The competent authority may then conduct inspections against the requirements of the relevant good practices before permitting such businesses to start trading. Manufacturers, importers and distributors of active substances will not only be subject to inspection on the basis of suspicions of non-compliance, but also on the basis of risk-analysis.
Authorised manufacturers of medicinal products who also manufacture and/or import active substances, either for use in their own products or products manufactured by other companies, are not exempt from the requirement to register.
Persons who are requested to import an active substance from a non-EEA country that provide facilities solely for transporting the active substance, or where they are acting as an import agent, imports the active substance solely to the order of another person who holds a certificate of good manufacturing practice issued by the licensing authority, are not required to register.
The registration regime for manufacturers, importers and distributors of active substances will be subject to an application procedure, followed by a determination procedure completed by MHRA.
The person applying for registration must notify MHRA immediately of any changes which have taken place as regards to the information in the registration form, where such changes may have an impact on quality or safety of the active substances that are manufactured, imported or distributed. These changes shall be treated as incorporated in the application form.
MHRA must grant or refuse an application for registration within 60 working days beginning immediately after the day on which a valid application is received.
MHRA will notify the applicant within 60 days of receipt of a valid application for registration whether they intend to undertake an inspection.
The applicant may not undertake any activity before either:
● 60 days have elapsed and the applicant has not been notified of the Agency’s intention to inspect, or
● following inspection the Agency has notified the applicant that they may commence their activities.
After inspection MHRA will prepare a report and communicate that report to the applicant. The applicant will have the opportunity to respond to the report. Within 90 days of an inspection MHRA shall issue an appropriate good practice certificate to the applicant, indicating that the applicant complies with the requirements of the relevant good practices. Where an applicant is found to be non-compliant with the requisite standards, a statement of non-compliance will be issued by MHRA.
If after 60 days of the receipt of the application form MHRA has not notified the applicant of their intention to carry out an inspection, the applicant may commence their business activity and regard themselves as registered. MHRA will issue a certificate to the applicant and enter the details into the Community Database .
This Community Database which is publicly available will enable competent authorities in other EEA Member States or other legal entities, to establish the bona fides and compliance of manufacturers, importers and distributors of active substances established in the UK and those in other EEA territories. MHRA will investigate concerns with regards to UK registrations of non-compliance and reciprocal arrangements will apply with other EEA Member States.
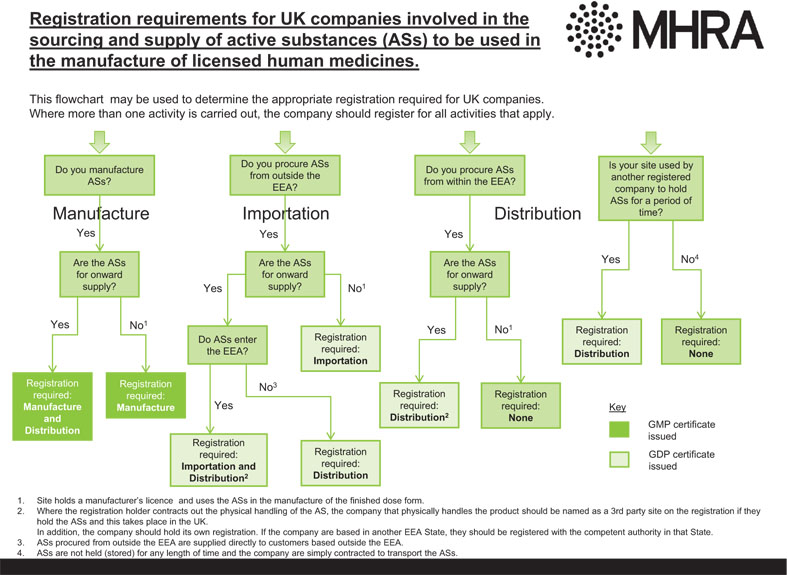
Figure 4.1 Registration requirements for UK companies involved in the sourcing and supply of active substances (ASs) to be used in the manufacture of licensed human medicines.
Conditions of Registration as a Manufacturer, Importer or Distributor of an Active Substance
A person in the UK may not import, manufacture or distribute, an active substance for use in a licensed human medicine unless they are registered with MHRA in accordance with the Human Medicines Regulations 2012 and the respective conditions of those Regulations are met.
Registration holders must submit to MHRA an annual update of any changes to the information provided in the application. Any changes which may have an impact on the quality or safety of the active substance which the registrant is permitted to handle must be notified to the Agency immediately.
An annual compliance report will need to be submitted:
● in relation to any application made before 31 March 2013, the date of application; and
● in relation to each subsequent reporting year, 30 April following the end of that year.
Where the Commission has adopted principles and guidelines of good manufacturing practice under the third paragraph of Article 471 of Directive 2001/83/EC which applies to an active substance manufactured in the UK, the registered manufacturer must comply with good manufacturing practice in relation to that active substance.
Where the Commission has adopted principles and guidelines of good distribution practice under the fourth paragraph of Article 47 of Directive 2001/83/EC which applies to an active substance distributed in the UK, the registered distributor must comply with good distribution practice in relation to that active substance.
Where the Commission has adopted principles and guidelines of good manufacturing practice under the third paragraph of Article 47 of Directive 2001/83/EC which applies to an active substance imported into the UK and where an active substance is imported from a third country the registered importer must comply with good distribution practice in relation to the active substance.
Under such circumstances the active substances must have been manufactured in accordance with standards which are at least equivalent to EU good manufacturing practice and when imported must be accompanied by a written confirmation from the competent authority of the exporting third country unless a waiver exists.
Directive 2001/83/EC has been amended to include a new definition of “active substance” which means any substance or mixture of substances intended to be used in the manufacture of a medicinal product and that, when used in its production, becomes an active ingredient of that product intended to exert a pharmacological, immunological or metabolic action with a view to restoring, correcting or modifying physiological functions or to make a medical diagnosis.
The manufacture of active substances should be subject to good manufacturing practice regardless of whether those active substances are manufactured in the Union or imported. Where active substances are manufactured in third countries it should be ensured that such substances have been manufactured to the relevant European standards of good manufacturing practice (GMP), so as to provide a level of protection of public health equivalent to that provided for by EU law.
A manufacturer or assembler of an active substance will have to comply with the principles and guidelines for GMP for active substances. Manufacture, in relation to an active substance, includes any process carried out in the course of making the substance and the various processes of dividing up, packaging, and presentation of the active substance. Assemble, in relation to an active substance, includes the various processes of dividing up, packaging and presentation of the substance, and “assembly” has a corresponding meaning. These activities will be the subject of a GMP certificate.
Importers of an active substance from a third country have to comply with the guidelines for Good Distribution Practice (GDP) in relation to the active substance. This activity will be the subject of a GDP certificate.
Distributors of an active substance within the UK which has been sourced from a manufacturer or an importer within the EU will have to comply with the guidelines for GDP for active substances. This activity will be the subject of a GDP certificate.
The 2001 Directive has been amended to permit the European Commission to adopt the following:
● the principles and guidelines of good manufacturing practice for active substances, by means of a delegated act; and
● the principles of good distribution practice for active substances, by means of adopted guidelines.
GMP for active substances is contained in Part II of the EU guidelines on Good Manufacturing Practice.
In February 2013 the European Commission consulted on its draft “Guidelines on the principles of good distribution practices for active substances for medicinal products for human use”.
The draft text sets out the quality system elements for the procuring, importing, holding, supplying or exporting active substances. However the scope of the proposed text excludes activities consisting of re-packaging, re-labelling or dividing up of active substances which are manufacturing activities and as such are subject to the guidelines on GMP of active substances. The draft text of the guidelines covers:
● Quality System
● Personnel
● Documentation
● Orders
● Procedures
● Records
● Premises and Equipment
● Receipt
● Storage
● Deliveries to Customers
● Transfer of Information
● Returns
● Complaints and Recalls
● Self-inspections.
The Falsified Medicines Directive modifies EU Medicines Directive 2001/83/EC and from 2 July 2013 introduces new requirements for active substances imported into the EEA for use in the manufacture of authorised medicinal products.
The Falsified Medicines Directive requires importers of active substances to obtain written confirmations from competent authorities in non-EEA countries (“third countries”) that the standards of manufacture of active substances at manufacturing sites on their territory are equivalent to EU good manufacturing practice (EU GMP). These confirmations are required before importation of active substances into the EU.
Each shipment of active substance received should be accompanied by a written confirmation from the competent authority of the exporting third country, stating that the active substance has been:
● manufactured to GMP standards at least equivalent to those laid down in the European Union;
● the third country manufacturing plant is subject to regular, strict and transparent inspections, and effective enforcement of GMP;
● in the event of non-conformance of the manufacturing site on inspection, such findings will be communicated to the European Union without delay.
The template for the written confirmation has been published in Part III of EudraLex, Volume 4 and is included in Chapter 2 of the Orange Guide.
Waiver from Written Confirmation
The Falsified Medicines Directive provides two waivers if written confirmations are not provided. The first is where the regulatory framework applicable to active substances in those third countries has been assessed by the European Commission (EC) as providing an equivalent level of protection of public health over active substance manufacture and distribution to those applied in the EU. This assessment follows a request from the exporting third country’s competent authority, and considers the regulatory framework for active substance manufacture and control and its equivalence to EU standards. Only if the exporting country is on the EC’s (“white”) list is the requirement for a written confirmation from that country’s competent authority removed.
The second waiver is where the third country active substance manufacturing site has been inspected by an EU Member State, has issued a certificate of compliance with EU-GMP and that it remains within its period of validity. This is an exceptional waiver intended to apply where it is necessary to ensure the availability of medicinal products. Member States using this waiver should communicate this fact to the European Commission in accordance with the legislation (Article 46b(4)). MHRA has notified the European Commission of its intent to use this waiver should that be necessary.
Procedure for Active Substance Importation
A summary of the overall active substance import process has been developed to promote a common expectation and common approaches by Member States and is available on the Heads of Medicines Agencies, website: http://www.hma.eu/43.html. The flow chart is reproduced here:
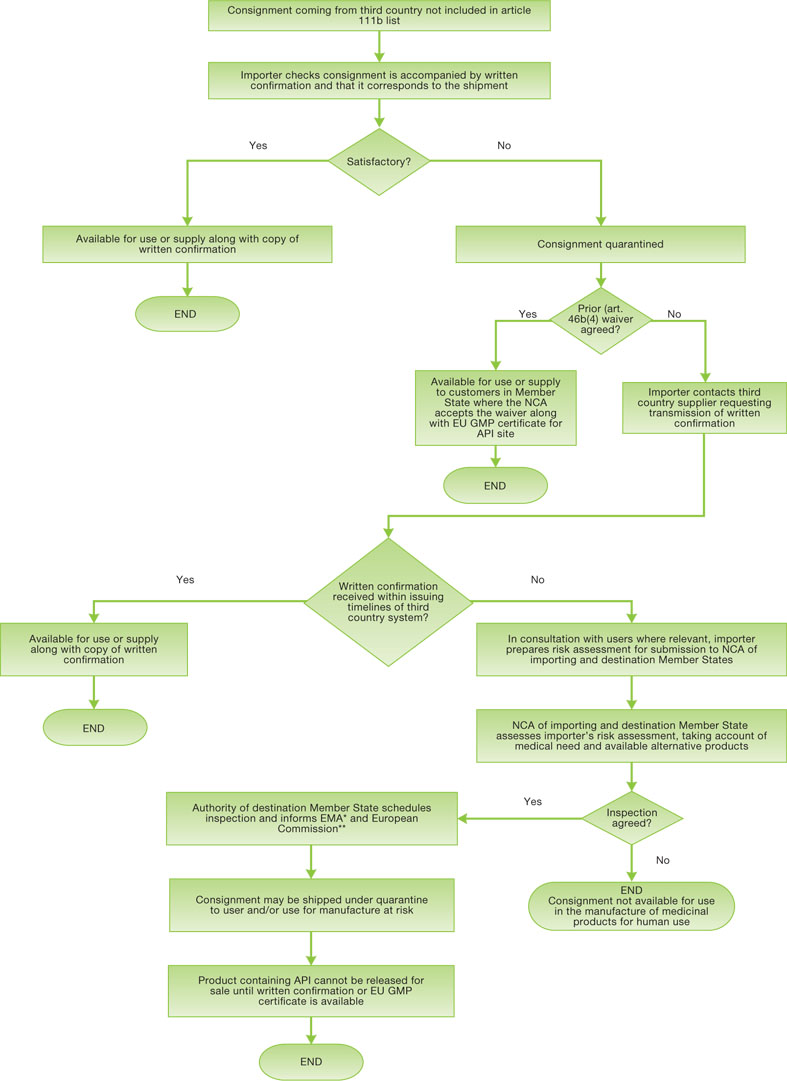
Figure 4.2 A flowchart of the overall active substance import process
*GMPINS@ema.europa.eu
**sanco-pharmaceuticals-d6@ec.europa.eu
Procedure for Waiver from Written Confirmation
UK based companies (registered importers and manufacturers who also import directly) who wish to import active substances under the second waiver should apply to gmpinspectorate@mhra.gsi.gov.uk using the form provided below which is available on the MHRA website.
Application Form for a waiver from the requirement to supply Written Confirmation with consignments of an imported Active Substance on the basis of a GMP certification of the Active Substance Manufacturer by an EEA Member State
A. Details of Third Country Manufacturing Site
Name of Active Substance: ____________________
(Note: Only one active substance permitted per Waiver application. Use INN nomenclature)
Name of Active Substance Manufacturer: _______________
Address of Active Substance Manufacturer: ______________
Country:______________
Third Country Competent Authority site/facility reference number (if known):___________
B. Reason for Application for this Waiver
The manufacturer/importer should attach a document explaining the reason for requesting this waiver. It should be noted that if the active substance is being sourced from a third country where the authority there is known to issue Written Confirmations then under normal circumstances it would be expected that a Written Confirmation would be the basis for importation of active substances from that country.
C. Details of GMP Certification
Any differences between the name and address supplied above and those details supplied on the GMP certificate must be justified in order to ensure efficient processing of the application.
Name of authority which issued the GMP certificate:_________________
Inspection Date Referenced on GMP certificate :_________________
Period of validity of GMP certificate* (if stated): _________________
(*this is 3 years unless there is a statement to the contrary)
Please attach a copy of the GMP certificate to this application form.
D. Details of Waiver Applicant
The Waiver Application may be submitted either by a site which has been registered with the MHRA for importation activities in the UK relating to active substances or by an authorised manufacturer / importer of human medicines in the UK which is using the imported active substance for manufacture of medicines for human use (excludes Investigational Medicinal Products) at its manufacturing address. If the importation activities (purchase of the active substance from a third country site or acting as the direct site of physical importation of the active substance) are being carried out directly by an authorised manufacturer of human medicines then both sections D1 and D2 below should be completed.
D1. Active Substance Importer
Active Substance Importers Registration No.:________________
Importation activity carried out for this active substance
(tick all that apply) ◽ Procurement (Purchasing) ◽ Site of physical importation
Registered Name of Importer :______________________
Registered Address of Importer:______________________
Country UK
D2. Authorised Manufacturer of Human Medicines using Imported Active Substance
Manufacturer’s / Importers Authorisation No.:______________________
Name of Authorised Manufacturer :______________________
Manufacturing Site Address :______________________
Country UK
E. Signature of Waiver Applicant
Signature:_________________ Date: _________________
Name (Print) :_________________
Position:_________________
F. Decision by MHRA on Waiver
Waiver Number:_________________
Waiver approved (Yes / No):_________________
Signature:_________________ Date:_________________
Name (Print):_________________
Position: _________________
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


