Study
Follow up duration
Type of intervention
% Patients with angina
BARI [1]
1 year
CABG, PCI
10 % CABG, 30 % PCI
COURAGE [2]
5 years
PCI, MT
26 % PCI, 28 %MT
RITA-2 [3]
1 year
PCI, MT
38 % PCI, 57 % MT
MASS-II [4]
1 year
CABG, PCI, MT
12 % CABG, 21 % PCI, 54 % OMT
FAME [5]
2 years
Angiography-guided PCI, FFR-guided PCI
24 % angiography- guided PCI, 20 % FFR-guided PCI
Revascularization procedures (either percutaneous or surgical) are performed with the aim of removing the flow limiting effect of epicardial coronary artery stenoses. This rationale is in line with the accepted pathophysiological mechanism of stable angina i.e., flow limiting epicardial stenosis, and the majority of patients undergoing revascularization benefit from the intervention. However, the situation is not straightforward as at least three main angina patient subgroups exist that deserve special consideration: (1) Patients who are deemed unsuitable for coronary revascularization because of diffuse coronary artery disease (CAD). Importantly, this subset represents an increasing population, particularly among subjects with diabetes and elderly patients; (2) Patients whose symptoms failed to be improved after percutaneous or surgical coronary procedures (recurrent angina). There are several possible causes for this i.e., bypass graft failure, restenosis, atherosclerotic disease progression and incomplete revascularization, among others. (3) Patients who undergo successful revascularization, and in whom none of the above mentioned mechanisms can be identified as a cause for symptom recurrence (persistent angina). In this latter group, factors other than epicardial stenosis, such as microvascular dysfunction, have been suggested as the underlying pathophysiological mechanism for persistent symptoms. While patients in the first subgroup experience a poor quality of life, subjects in the latter two experience the additional burden of having to undergo repeat invasive procedures, which leads to increased patient and physician frustration and, last but not least, increased healthcare costs. Irrespective of the underlying cause, the final common pathophysiological mechanism is a reduced myocardial oxygen supply which is resistant to revascularization and traditional medical therapies.
Over the last 30 years considerable progress has been made in the therapeutic options for ischemic heart disease (IHD). Among these, metabolic modulation therapy, using agents such as trimetazidine, has been shown to confer significant symptom relief. The drug can be safely added to ongoing therapy with β-blockers (BBs), calcium channel blockers (CCBs), and nitrates, and has not been shown to display unfavorable drug interactions [8].
Cardiac Metabolism and the Rationale for Metabolic Modulation Therapy
In the adult heart, glycolysis and mitochondrial oxidative metabolism are the principal sources of energy production, with the later accounting for more than 95 % of the total amount required for heart function. Under normal conditions, 50–70 % of the energy produced by mitochondrial oxidative metabolism is derived from fatty acid β-oxidation. The remaining 30–50 % is achieved from the oxidative metabolism of the glycolytic products of glucose (pyruvate) (Fig. 8.1).
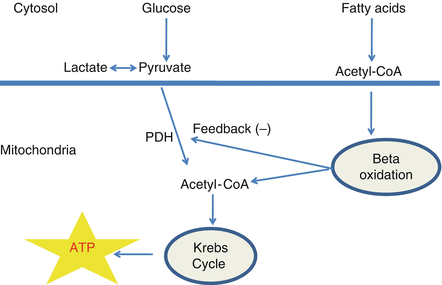
Figure 8.1
Schematic representation of main ATP sources in normal conditions (see text for details)
Relevance of Myocardial Metabolic Alterations in IHD
In aerobic conditions, fatty acid oxidation (FAO) yields more adenosine triphosphate (ATP) per gram of substrate than glucose oxidation (GO). However, FAO requires about 10–15 % more oxygen to produce an equivalent amount of ATP, thus representing a less efficient energy production pathway [9].
Such a property becomes particularly relevant in of the presence of a reduced oxygen supply such as in flow limiting IHD. Glucose metabolism begins with glycolysis, a cytosolic process that converts glucose to pyruvate. Glycolysis supplies pyruvate to the enzyme pyruvate dehydrogenase (PDH), which is the rate limiting enzyme for GO [9].
When PDH is active, pyruvate enters the mitochondria and is converted to acetyl-CoA, therefore fueling the Krebs cycle for ATP production. Conversely, FAO, which occurs within the mitochondria, is another source of acetyl-CoA for the Krebs cycle. These two sources of acetyl-CoA are highly dependent on and affected from each other, a phenomenon known as the ‘Randle cycle’ [10].
In the presence of large amounts of fatty acids, FAO is stimulated, while GO is directly inhibited through PDH activity [11].
As a consequence, pyruvate cannot enter the mitochondria and therefore cannot fuel oxidative metabolism. This results in the production of only two ATP for each glucose molecule (versus 36 ATP during GO) and conversion of pyruvate to lactate with resultant proton accumulation.
As a consequence of ischemia, less ATP is generated within mitochondria. This triggers accelerated glycolysis and reduces cell pH, leads to calcium accumulation, potassium efflux and adenosine formation [12].
Of interest, pain resulting from myocardial ischemia (angina) is associated with an enhanced catecholamine release and increased lipolysis. This condition is also associated with an increase in circulating fatty acids levels, a relative increase in FAO and therefore (through the ‘Randle cycle’) a reduced GO rate.
Myocardial ischemia has therefore a prominent role in increasing the uncoupling between glycolysis and GO by contemporaneously increasing glycolysis and reducing GO rates, further reducing cardiac efficiency. The need to use ATP for reestablishing ionic homeostasis instead of supporting contractile function is another cause for reduced cardiac efficiency. In addition, intracellular proton accumulation also directly decreases the efficiency of the contractile proteins and therefore cardiac efficiency [13].
Therefore, therapeutic interventions aimed at shifting myocardial substrate utilization from fatty acid towards glucose metabolism would particularly benefit cardiac efficiency and IHD symptoms. Given the interdependence between FAO and GO oxidation this can be achieved by either inhibiting FAO or stimulating GO. In this chapter we will focus on the use of trimetazidine, a partial inhibitor of FAO, in patients with chronic stable angina.
Beneficial Cellular Effects of Fatty Acid Oxidation Inhibition by Trimetazidine
There are numerous ways of inhibiting cardiac FAO, some of which include the inhibition of fatty acid transport into the cardiac myocyte, the inhibition of fatty acid uptake into the mitochondria, and the inhibition of the enzymatic machinery of the β-oxidative pathway itself.
Trimetazidine (1-[2,3,4-trimethoxybenzyl] piperazine dihydrochloride), a metabolic modulator agent that has been used for more than two decades in Europe, inhibits FAO by blocking the β-oxidative enzyme, long-chain 3-ketoacyl CoA thiolase (LC 3-KAT) [14].
This effect results in an increase in PDH activity which compensates the reduced availability of acyl-CoA derived from β-oxidation of fatty acids, by providing them through GO. In this way, stimulation of GO increases glycolysis/glucose oxidation coupling, resulting in a decreased proton production, and a decreased both intracellular calcium overload and free radical production. Finally, these chain effects prevent a significant decrease in ATP and phosphocreatine levels in response to hypoxia or ischemia, preserve ionic pump function and therefore translate into an improved cardiac efficiency and reduced symptoms. Importantly, these effects are not associated with significant alterations in hemodynamic parameters, a fact of particular important for patients who receive anti-ischemic treatment with hemodynamic active agents [14] (Fig. 8.2).
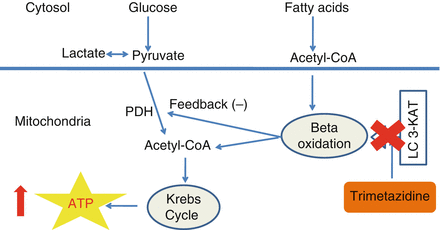
Figure 8.2
Trimetazidine inhibits LC-3KAT, one of the enzymes of fatty acid oxidation. In this way there is a reduced production of acetyl-CoA originating from fatty acid oxidation and therefore a relief of PDH activity. This results in increased glucose oxidation and, therefore a higher myocardial efficiency. Abbreviations: ATP (adenosine triphosphate)
Novel Evidence on the Effects of Trimetazidine on Endothelial Function
Accumulating evidence suggests that trimetazidine has the ability to improve endothelium-dependent relaxation through direct and/or indirect effects on the endothelium itself. Experimental and clinical studies have shown that patients with greater oxidative stress and high oxygen free radical production have severe endothelial dysfunction due to high plasma levels of malondialdehyde and lipid hydroperoxides [15].
Trimetazidine improves endothelium-dependent vasodilation, and this effect is correlated with decreased plasma levels of both malondialdehyde and hydroperoxides. Using flow-mediated dilation (FMD) techniques, Park et al. evaluated the effects of sheath induced injury and trimetazidine on endothelial function of the radial artery (RA) after transradial coronary artery angiography (TRCAG) or transradial percutaneous coronary intervention (TRPCI) [16].
Ten weeks after the procedure, FMD was reduced in the control group, whereas no difference was found in the trimetazidine group, suggesting that treatment with trimetazidine improves endothelial function of the cannulated RA.
Following PCI, inflammation, thrombosis, cellular proliferation, and extracellular matrix production, contribute to neo-intimal hyperplasia and post-procedural luminal narrowing leading to restenosis and delayed re-endothelialization, all of which represent important clinical problems. Yoon at al showed that 4 weeks of trimetazidine treatment resulted in a dose-dependent reduction in the intima-media thickness ratio of the carotid artery of type 1 and 2 rat model after balloon injury [17].
This effect was accompanied by decreased proliferation of vascular smooth muscle cells (VSMCs) and accelerated re-endothelialization after carotid balloon injury. In vitro experiments with trimetazidine showed decreased VSMCs proliferation and migration, whereas human umbilical vein endothelial cells (HUVECs) displayed increased proliferation and decreased apoptosis. Antioxidative effects of trimetazidine were observed in both VSMCs and HUVECs. The authors concluded that reduction of restenosis by trimetazidine treatment was the net effect of changes in antioxidative and anti-inflammatory properties, which are cell-specific effects on either survival or apoptosis [17].
In line with these results, Chen et.al. recently published the results of a randomized study involving 786 patients who underwent drug eluting stent placement for chronic IHD. At 1 year from the index procedure, treatment with trimetazidine on top of optimal medical therapy significantly reduced in-stent-restenosis and major cardiac adverse events [18].
Clinical Efficacy of Trimetazidine
Treatment with trimetazidine has been shown to confer beneficial effects in various clinical scenarios including chronic stable angina, acute coronary syndromes (ACS), as well as heart failure. In patients with chronic angina, trimetazidine increases exercise capacity and delays the appearance of symptoms and ECG changes during exercise [15, 19, 20]. Importantly, the benefits observed after acute administration are maintained in long term treatment, which is well tolerated by patients [21]. Trimatazidine has no significant negative inotropic effects or vasodilator properties either at rest or during dynamic exercise [22]. The efficacy of trimetazidine as an anti-angina drug has been assessed in randomized, placebo-controlled studies, both as ‘solo’ treatment and in combination or comparison with BBs and CCBs (Table 8.2).
Table 8.2
Main trimetazidine studies considered in this review
Author | Year | Study title |
|---|---|---|
Trimetazidine as monotherapy | ||
Sellier et al. [20] | 1986 | The effects of trimetazidine on ergometric parameters of exercise-induced angina. |
Passeron et al. [21] | 1994 | Effectiveness of trimetazidine in stable effort angina due to chronic coronary insufficiency. |
Trimetazidine with B-blockers | ||
Szwed et al. [23] | 2001 | Combination treatment in stable effort angina using trimetazidine and metoprolol. TRIMPOL II study. |
Trimetazidine with CCBs | ||
Monpere et al. [24] | 1990 | Combination of trimetazidine with nifedipine in effort angina. |
Manchanda et al. [25] | 1997 | Combination treatment with trimetazidine and diltiazem in stable angina pectoris. |
Trimetazidine versus nitrates | ||
Hanania et al. [26] | 2002 | Comparison between trimetazidine and mononitrate isosorbide for patients receiving B-blockers. |
Trimetazidine versus B-blockers | ||
Detry et al. [27] | 1994 | Trimetazidine: a new concept in the treatment of angina. Comparison with propranolol in patients with stable angina. |
Trimetazidine versus CCBs | ||
Dalla-Volta et al. [28] | 1990 | Comparison of trimetazidine with nifedipine in effort angina: a double-blind, crossover study.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|