Treatment of Primary Sclerosing Cholangitis
David B. Adams
Katherine A. Morgan
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease of uncertain etiology, characterized by inflammation and fibrosis of the intrahepatic and extrahepatic bile ducts. PSC is an indolent disease that progresses to biliary cirrhosis in most patients. PSC is rare, occurring in 8.5 to 13.6 per 100,000 persons in the United States. Whites and Northern European men are affected more commonly than women (men-to-women ratio is approximately 2:1) and the mean age at diagnosis is 39 years. The majority of cases of PSC occur in patients with inflammatory bowel disease (IBD). Although immunopathologic mechanisms are implicated in PSC, it is not a typical autoimmune disease and responds poorly to steroids and other immunosuppressive therapies.
PSC confers an increased risk of cholangiocarcinoma (CCA) as well as other gastrointestinal malignancies. There is a shortened life expectancy with PSC, with a median survival after diagnosis between 9 and 18 years. Patients with PSC have a diminished health-related quality of life compared with the general population. Medical and surgical therapies have had disappointing results in the alteration of the natural history of PSC, with liver transplantation offering the only life-extending therapeutic option. The focus of current interventions is to manage associated complications rather than to alter disease progression. The surgical management of PSC warrants consideration in any discussion about benign and malignant biliary tract disorders. The role of endoscopic therapies, the value and conduct of extrahepatic biliary resection (EHBR), and the indications for and timing of liver transplantation remain lively and controversial topics.
Pathogenesis
The cause of PSC is unknown and is most likely multifactorial. Current implicated factors include immunopathogenetic mechanisms and an inflammatory response to infectious agents.
Evidence of a genetic component is demonstrated in the familial predisposition to PSC, with a 100-fold increased risk of disease amongst siblings. A distinguishable pattern of inheritance, however, is not apparent. PSC is associated with certain human leukocyte antigen (HLA) haplotypes, most commonly HLA-B8 and HLA-DR3. Additionally, HLA-DR2, which is found in patients with a younger onset of disease, and HLA-DR4, which is a marker for more rapid disease progression, are associated with PSC. PSC is linked with other autoimmune disorders, including IBD, thyroiditis (8%), type I diabetes (10%), and psoriasis (4%). Patients with PSC are noted to have hypergammaglobulinemia with elevated IgM and alterations in activation of the complement pathway. Studies have also shown a decreased number of circulating T cells with increased T cells in liver infiltrates, and an increase in the CD4:CD8 ratio.
Eighty percent of patients with PSC have elevated perinuclear antineutrophil cytoplasmic antibodies (pANCA). Up to 97% of patients with PSC demonstrate at least one autoantibody. Given current evidence, some immunogenetic mechanism seems likely to be central to the development of PSC.
Multiple infectious agents in various roles have been proposed as potential instigators of the inflammatory response resulting in PSC. Chronic portal bacteremia or bacterial metabolites from gut flora, particularly in patients with IBD, have been cited. Potentially these factors activate hepatic Kupffer cells, leading to the production of tumor necrosis factor, an inflammatory response, and ultimately biliary fibrosis. Alternatively, bacterial antigens may behave as a molecular mimic of an autoantigen, stimulating an autoimmune response. In addition, chronic viral infections such as cytomegalovirus have been investigated. While such environmental processes may certainly play a role, the exact mechanism is not yet understood.
Associated Diseases
Inflammatory Bowel Disease
The majority of patients with PSC suffer from IBD, which typically appears prior to PSC. Approximately 63% to 80% of PSC patients have ulcerative colitis (UC); another 5% to 13% suffer from Crohn’s disease. Conversely, 2% to 7.5% of UC patients and 1.4% to 3.4% of Crohn’s patients have PSC. The correlation between IBD and PSC is so strong that PSC outside the setting of IBD may indeed be a separate clinical entity.
The indications and timing of colectomy in patients with PSC and UC is controversial. In general, patients with UC and PSC in whom colectomy is indicated do well to avoid an ileostomy, given the risk for stomal varices, which can be a source of problematic morbidity. A restorative proctocolectomy with ileoanal anastomosis is the preferred surgical option.
Liver transplantation for PSC-related cirrhosis may alter the course of UC to a more aggressive disease. Alternatively, in patients with advanced hepatic disease, a colectomy prior to transplantation may result in hepatic decompensation. For this reason, pretransplant proctocolectomy is currently reserved for patients with appropriate clinical indications (duration of colitis greater than 10 years, pancolitis, evidence of colonic dysplasia).
Patients with PSC and UC have been reported to be at higher risk for colorectal cancer than patients with UC alone. In one study, the cumulative risk of developing colorectal cancer in patients with PSC and UC after liver transplantation was 14% at 5 years. These patients therefore warrant close surveillance and early consideration for colectomy, particularly if they develop dysplasia or have a long duration of clinically active colitis.
Biliary Malignancy
CCA, the most lethal complication of PSC, occurs in 6% to 23% of patients with PSC. The diagnosis of CCA is very difficult in the setting of PSC, given the stricturing pattern of the disease and the poor sensitivity of brush biopsies. Diagnosis may be illusive until later stages of malignancy. Patients with PSC and CCA are typically younger at diagnosis than patients with bile duct cancers without PSC.
The role of serum CA19-9 levels as a screening mechanism for CCA in patients with PSC is unclear. A recent study purports that a CA19-9 level greater than 129 U/mL offers a sensitivity of 78.6% and specificity of 98.5% for CCA in the setting of PSC. Unfortunately, CA19-9 levels are a poor screening test, as their utility is chiefly in identifying patients with more advanced stage malignancies.
Magnetic resonance cholangiography (MRC) as well as traditional cholangiography are not adequate to distinguish benign PSC-related biliary strictures from the malignant strictures of CCA. Endoscopic brushings are
highly specific, but lack sensitivity. In general, high suspicion for malignancy should be maintained surrounding the development of any dominant stricture, particularly one that is unresponsive to endoscopic therapies or is associated with a decline in the patient’s clinical condition.
highly specific, but lack sensitivity. In general, high suspicion for malignancy should be maintained surrounding the development of any dominant stricture, particularly one that is unresponsive to endoscopic therapies or is associated with a decline in the patient’s clinical condition.
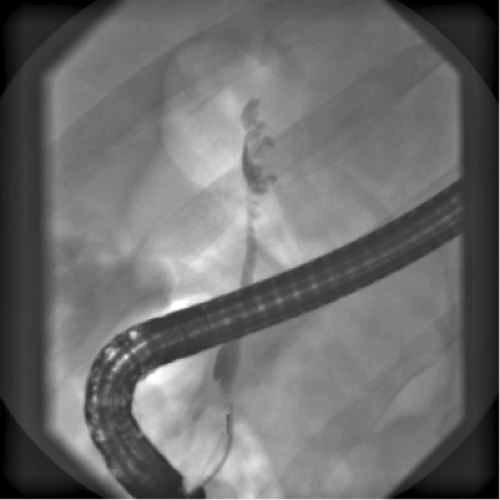 Fig. 1. ERC image typical of PSC, with intermittent stricturing of the common bile duct creating a beaded appearance. |
CCA in the setting of PSC has a very poor outcome, with a median survival of 5 to 11 months. At most centers, known CCA, even localized, precludes transplant due to poor expected survival, reported as 35% at 5 years following transplant. Some centers, however, have proposed neoadjuvant therapy followed by transplantation for early, isolated CCA, citing 45% long-term survival.
Hepatocellular cancer (HCC) is also seen in patients with advanced liver disease secondary to PSC. As in patients with liver failure from other causes, selected patients with limited HCC (i.e., Milan criteria: single tumor, less than 5-cm diameter, or three or fewer tumor nodules, less than 3-cm diameter) can expect reasonable survival rates, as high as 85% at 4 years, after liver transplantation for HCC.
Gallbladder cancer, as well, has an increased incidence in patients with PSC. Accordingly, gallbladder polyps should prompt due concern and consideration for cholecystectomy in these patients.
Fifteen to fifty-five percent of patients with PSC are asymptomatic at presentation, typically discovered by abnormal hepatic enzyme profiles. When present, symptoms include abdominal pain (20% to 37%), pruritus (10% to 40%), jaundice (6% to 30%), fever (4% to 30%), diarrhea (8%), and fatigue (6%). Approximately 10% of patients present with bacterial cholangitis and 4% present with variceal bleeding due to portal hypertension. A changing trend in the pattern of presentation at diagnosis has been noted recently, with an increase in patients who are asymptomatic, older, and do not have IBD. Physical examination is typically unremarkable at presentation, although those with more advanced disease may have organomegaly, hyperpigmentation, and other clinical signs of advanced liver dysfunction.
Patients generally present with biochemical markers of cholestasis. Elevated alkaline phosphatase and gamma-glutamyl transpeptidase are consistent, with values generally greater than three times the normal. Lesser elevations in the transaminases can also be seen. The majority of patients (greater than 60%) will have a normal bilirubin, with elevated bilirubin a potential herald of advanced disease, biliary stricture, choledocholithiasis, cholangitis, or CCA. It is notable that the liver biochemistries may fluctuate through the course of the disease.
pANCA are present in 80% of patients but are not specific for PSC. Other autoantibodies including antinuclear antibodies and antismooth muscle antibodies may be present as well. Antimitochondrial antibodies are not typical of PSC and can help in differentiating the disease from primary biliary cirrhosis.
Radiography
Cholangiography is the primary medium of diagnosis of PSC. Classic features include multifocal, diffusely distributed strictures of the biliary system, interspersed with short intervening segments of normal or dilated bile ducts, to produce a “string of beads” appearance. In the vast majority of patients the intrahepatic and extrahepatic ducts are involved, although in a minority of cases, the disease may appear isolated to the intrahepatic ducts, or even less commonly the extrahepatic ducts alone. A notable, rare exception is the so-called small duct variant of PSC. In this presentation, involved ducts are presumed too small to be demonstrated on cholangiography, and patients are typically IBD patients identified by abnormalities in their liver biochemistries in conjunction with consistent findings at liver biopsy.
Endoscopic retrograde cholangiography (ERC) is the procedure of choice in diagnosis and evaluation of the biliary ductal anatomy (Fig. 1). Complications have been reported in 3% to 10% of patients undergoing ERC for PSC. There is an increased risk of cholangitis after endoscopic intervention, and antibiotic prophylaxis periprocedurally is recommended. When ERC is not feasible, percutaneous transhepatic cholangiography (PTC) can be undertaken.
MRC has been used increasingly in the evaluation and management of biliary diseases (Fig. 2). As a noninvasive study, MRC avoids the potential complications of ERC. In PSC, MRC has been shown to be accurate and comparable with ERC. MRC has the advantage of allowing visualization of the biliary tree proximal to tight strictures as well as the hepatic parenchyma, both of which are not well evaluated by ERC. MRC, however, is not as sensitive for disease in small peripheral ducts and does not allow for intervention with brushings, dilation, or stenting. Certainly, MRC can be a valuable modality in initial diagnosis of PSC, although its exact role in later management of disease is unclear.
Histology
PSC is characterized histologically by concentric periductal fibrosis, known as “onion skinning,” duct obliteration, and ductopenia. These classic histologic findings may be illusive on liver biopsy due to sampling error, and may be found in as few as 15% of patients. Other less specific but consistent
histologic findings include pseudoxanthomatous changes, copper deposition, and Mallory bodies. A histologic staging system has been described, with stage I showing cholangitis and periportal hepatitis, stage II consisting of periportal fibrosis or periportal hepatitis, stage III showing septal fibrosis or bridging necrosis, and stage IV demonstrating biliary cirrhosis. Liver biopsy is most useful in cases with challenging diagnosis, such as in patients with suspected small duct PSC, without definitive cholangiographic evidence of disease.
histologic findings include pseudoxanthomatous changes, copper deposition, and Mallory bodies. A histologic staging system has been described, with stage I showing cholangitis and periportal hepatitis, stage II consisting of periportal fibrosis or periportal hepatitis, stage III showing septal fibrosis or bridging necrosis, and stage IV demonstrating biliary cirrhosis. Liver biopsy is most useful in cases with challenging diagnosis, such as in patients with suspected small duct PSC, without definitive cholangiographic evidence of disease.
Immunoglobulin subtype G4-related sclerosing cholangitis has been recognized in the past decade. This disease process is related to autoimmune pancreatitis, and appears to be a distinct disease from PSC. Patients with IgG4-related sclerosing cholangitis have elevated serum IgG4 levels, and on histology show lymphoplasmacytic infiltration and infiltration with IgG4-containing plasma cells in the biliary system. Unlike PSC, IgG4-related sclerosing cholangitis is often responsive to steroid therapy.
Natural History of the Disease
While many patients with PSC are asymptomatic at diagnosis, all develop symptoms (abdominal pain, pruritus, jaundice, fatigue) with time and ultimately biliary cirrhosis. Spontaneous regression or resolution has not been described. The time course of progression is variable. The median survival from diagnosis of PSC to death or transplant is between 9 and 18 years. Patients who are asymptomatic on presentation seem to have a better prognosis, although up to 17% may have cirrhosis at the time of diagnosis despite the lack of clinical signs. Disease progression of symptomatic patients is typically more aggressive. During a mean follow-up of 6 years, 41% to 49% of symptomatic patients die or require liver transplantation.
Prognostic models to aid in clinical decision making in PSC, particularly with regard to timing of liver transplantation, have been developed. The most commonly used is the multicenter model, based on the course of disease in 486 patients from 3 centers, which incorporates patient age, total bilirubin, histologic stage, and the presence of splenomegaly. More recently developed is the revised Mayo model, which assimilates patient age, total bilirubin, aspartate aminotransferase, albumin, and occurrence of variceal bleeding, and does not require histology. Once more advanced liver failure develops, the Model for End-Stage Liver Disease (MELD) score is appropriately applied, particularly in determining suitability and priority in liver transplantation.
Since the pathogenesis of PSC is poorly understood, development of effective medical therapies is challenging. Many unsuccessful medical therapies for PSC have been evaluated, including penicillamine, methotrexate, cyclosporine, mycophenolate mofetil, infliximab, and several others. PSC has not yet proven responsive to immunosuppressive regimens.
Ursodeoxycholic acid (UDCA) is the most common and best-studied medical therapy utilized in the management of PSC. UDCA is a hydrophilic bile acid, representing 3% of the normal bile acid pool, and has been employed in the management of various cholestatic diseases. In therapeutic settings, UDCA acts primarily as a choleretic, increasing bile flow. It also has gastrointestinal cytoprotective effects, immune-modulating effects, and inhibitory effects on apoptosis in PSC. UDCA in standard dosing (8 to 15 mg/kg/d) has been shown to improve liver biochemistries and to improve the histologic staging of PSC, although no effect on survival has been shown. Given these favorable results, UDCA in higher doses has been evaluated. Multiple pilot studies and one large multicenter study from Europe evaluating high-dose UDCA (23 to 30 mg/kg/d) have shown improved liver function tests, histology, and cholangiographic appearance of PSC. In addition, a trend toward improved survival with therapy was seen though not statistically significant. Improved observed survival as compared with expected survival utilizing the Mayo risk score was noted. However, a recent multicenter, prospective double-blind, controlled study with long-term follow-up evaluating high-dose UDCA has identified improvement in hepatic enzyme profiles in all treated patients, but this study required early termination due to increased risk for adverse outcome including death and time to liver transplantation in the treatment group, particularly in patients with more advanced disease.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


