Figure 2-1. The molecular targets of psychotropic drugs. There are only a few major sites of action for the wide expanse of psychotropic drugs utilized in clinical practice. Approximately one-third of psychotropic drugs target one of the twelve-transmembrane-region transporters for a neurotransmitter (A), while another third target seven-transmembrane-region receptors coupled to G proteins (B). The sites of action for the remaining third of psychotropic drugs include enzymes (C), four-transmembrane-region ligand-gated ion channels (D), and six-transmembrane-region voltage-sensitive ion channels (E).
Neurotransmitter transporters as targets of drug action
Classification and structure
Neuronal membranes normally serve to keep the internal milieu of the neuron constant by acting as barriers to the intrusion of outside molecules and to the leakage of internal molecules. However, selective permeability of the membrane is required to allow discharge as well as uptake of specific molecules, to respond to the needs of cellular functioning. Good examples of this are neurotransmitters, which are released from neurons during neurotransmission and, in many cases, are also transported back into presynaptic neurons as a recapture mechanism following their release. This recapture – or reuptake – is done in order for neurotransmitter to be reused in a subsequent neurotransmission. Also, once inside the neuron, most neurotransmitters are transported again into synaptic vesicles for storage, protection from metabolism, and immediate use during a volley of future neurotransmission.
Both types of neurotransmitter transport – presynaptic reuptake as well as vesicular storage – utilize a molecular transporter belonging to a “superfamily” of twelve-transmembrane-region proteins (Figures 2-1A and 2-2). That is, neurotransmitter transporters have in common the structure of going in and out of the membrane 12 times (Figure 2-1A). These transporters are a type of receptor that binds to the neurotransmitter prior to transporting that neurotransmitter across the membrane.
Recently, details of the structures of neurotransmitter transporters have been determined; this has led to a proposed subclassification of neurotransmitter transporters. That is, there are two major subclasses of plasma membrane transporters for neurotransmitters. Some of these transporters are presynaptic and others are on glial membranes. The first subclass consists of sodium/chloride-coupled transporters, called the solute carrier SLC6 gene family, and includes transporters for the monoamines serotonin, norepinephrine, and dopamine (Table 2-1 and Figure 2-2A) as well as for the neurotransmitter GABA (gamma-aminobutyric acid) and the amino acid glycine (Table 2-2 and Figure 2-2A). The second subclass consists of high-affinity glutamate transporters, also called the solute carrier SLC1 gene family (Table 2-2 and Figure 2-2A).
| Transporter | Common abbreviation | Gene family | Endogenous substrate | False substrate |
|---|---|---|---|---|
| Serotonin transporter | SERT | SLC6 | Serotonin | Ecstasy (MDMA) |
| Norepinephrine transporter | NET | SLC6 | Norepinephrine | Dopamine |
| Epinephrine | ||||
| Amphetamine | ||||
| Dopamine transporter | DAT | SLC6 | Dopamine | Norepinephrine |
| Epinephrine | ||||
| Amphetamine |
MDMA, 3,4-methylenedioxymethamphetamine
| Transporter | Common abbreviation | Gene family | Endogenous substrate |
|---|---|---|---|
| GABA transporter 1 (neuronal and glial) | GAT1 | SLC6 | GABA |
| GABA transporter 2 (neuronal and glial) | GAT2 | SLC6 | GABA β-alanine |
| GABA transporter 3 (mostly glial) | GAT3 | SLC6 | GABA β-alanine |
| GABA transporter 4, also called betaine transporter (neuronal and glial) | GAT4 | SLC6 | GABA betaine |
| BGT1 | |||
| Glycine transporter 1 (mostly glial) | GlyT1 | SLC6 | Glycine |
| Glycine transporter 2 (neuronal) | GlyT2 | SLC6 | Glycine |
| Excitatory amino acid transporters 1–5 | EAAT1–5 | SLC1 | L-glutamate |
| L-aspartate |
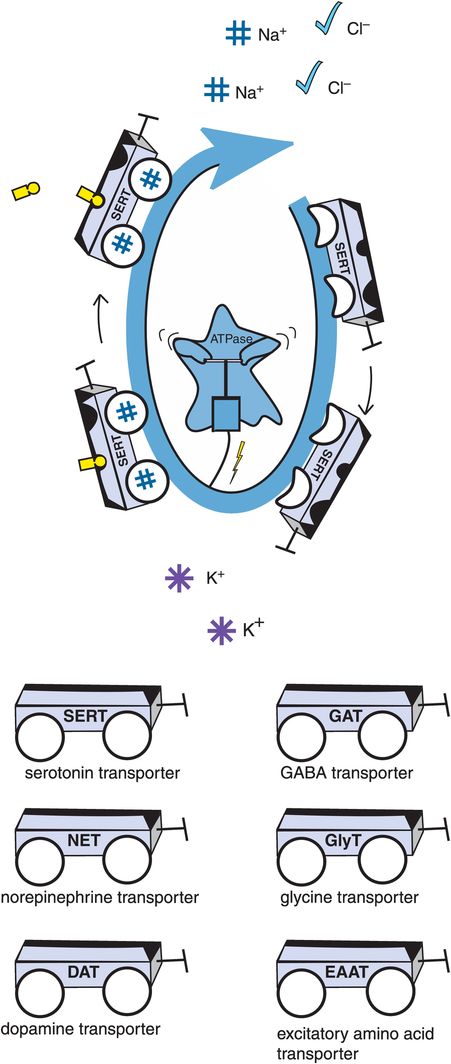
A. Sodium-potassium ATPase. Transport of many neurotransmitters into the presynaptic neuron is not passive, but rather requires energy. This energy is supplied by sodium-potassium ATPase (adenosine triphosphatase), an enzyme that is also sometimes referred to as the sodium pump. Sodium-potassium ATPase continuously pumps sodium out of the neuron, creating a downhill gradient. The “downhill” transport of sodium is coupled to the “uphill” transport of the neurotransmitter. In many cases this also involves cotransport of chloride and in some cases countertransport of potassium. Examples of neurotransmitter transporters include the serotonin transporter (SERT), the norepinephrine transporter (NET), the dopamine transporter (DAT), the GABA transporter (GAT), the glycine transporter (GlyT), and the excitatory amino acid transporter (EAAT).
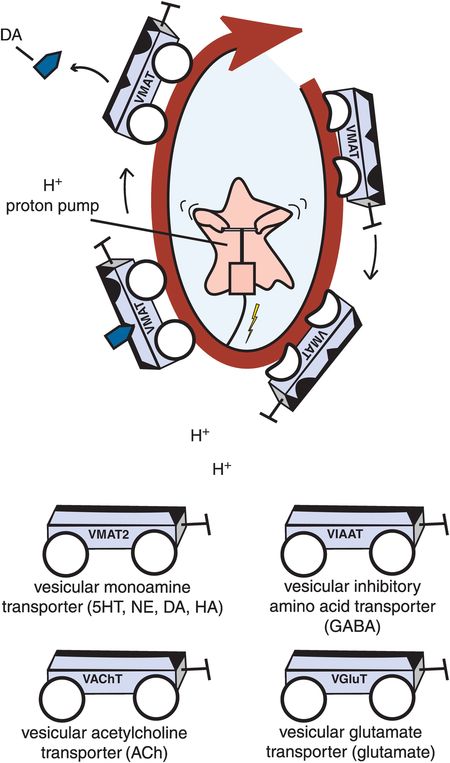
B. Vesicular transporters. Vesicular transporters package neurotransmitters into synaptic vesicles through the use of a proton ATPase, or proton pump. The proton pump utilizes energy to pump positively charged protons continuously out of the synaptic vesicle. Neurotransmitter can then be transported into the synaptic vesicle, keeping the charge inside the vesicle constant. Examples of vesicular transporters include the vesicular monoamine transporter (VMAT2), which transports serotonin, norepinephrine, dopamine, and histamine; the vesicular acetylcholine transporter (VAChT), which transports acetylcholine; the vesicular inhibitory amino acid transporter (VIAAT), which transports GABA; and the vesicular glutamate transporter (VGluT), which transports glutamate.
In addition, there are three subclasses of intracellular synaptic vesicle transporters for neurotransmitters. The SLC18 gene family comprises the vesicular monoamine transporters (VMATs) for serotonin, norepinephrine, dopamine, and histamine and the vesicular acetylcholine transporter (VAChT). The SLC32 gene family consists of the vesicular inhibitory amino acid transporters (VIAATs). Finally, the SLC17 gene family consists of the vesicular glutamate transporters, such as VGluT1–3 (Table 2-3 and Figure 2-2B).
| Transporter | Common abbreviation | Gene family | Endogenous substrate |
|---|---|---|---|
| Vesicular monoamine transporters 1 and 2 | VMAT1 | SLC18 | Serotonin |
| VMAT2 | Norepinephrine | ||
| Dopamine | |||
| Vesicular acetylcholine transporter | VAChT | SLC18 | Acetylcholine |
| Vesicular inhibitory amino acid transporter | VIAAT | SLC32 | GABA |
| Vesicular glutamate transporters 1–3 | VGluT1–3 | SLC17 | Glutamate |
Monoamine transporters (SLC6 gene family) as targets of psychotropic drugs
Reuptake mechanisms for monoamines utilize unique presynaptic transporters (Figure 2-2A) but the same vesicular transporter in all three monoamine neurons (histamine neurons also use the same vesicular transporter) (Figure 2-2B). That is, the unique presynaptic transporter for serotonin is known as SERT, for norepinephrine is known as NET, and for dopamine is known as DAT (Table 2-1 and Figure 2-2A). All three of these monoamines are then transported into synaptic vesicles of their respective neurons by the same vesicular transporter, known as VMAT2 (vesicular monoamine transporter 2) (Figure 2-2B and Table 2-3).
Although the three presynaptic transporters – SERT, NET, and DAT – are unique in their amino acid sequences and binding affinities for monoamines, each presynaptic monoamine transporter nevertheless has appreciable affinity for amines other than the one matched to its own neuron (Table 2-1). Thus, if other transportable neurotransmitters or drugs are in the vicinity of a given monoamine transporter, they may also be transported into the presynaptic neuron by hitchhiking a ride on certain transporters that can carry them into the neuron.
For example, the norepinephrine transporter (NET) has high affinity for the transport of dopamine as well as for norepinephrine, the dopamine transporter (DAT) has high affinity for the transport of amphetamines as well as for dopamine, and the serotonin transporter (SERT) has high affinity for the transport of “ecstasy” (the drug of abuse MDMA or 3,4-methylenedioxymethamphetamine) as well as for serotonin (Table 2-1).
How are neurotransmitters transported? Monoamines are not passively shuttled into the presynaptic neuron, because it requires energy to concentrate monoamines into a presynaptic neuron. That energy is provided by transporters in the SLC6 gene family coupling the “downhill” transport of sodium (down a concentration gradient) with the “uphill” transport of the monoamine (up a concentration gradient) (Figure 2-2A). Thus, the monoamine transporters are really sodium-dependent cotransporters; in most cases, this involves the additional cotransport of chloride, and in some cases the countertransport of potassium. All of this is made possible by coupling monoamine transport to the activity of sodium-potassium ATPase (adenosine triphosphatase), an enzyme sometimes called the “sodium pump” that creates the downhill gradient for sodium by continuously pumping sodium out of the neuron (Figure 2-2A).
The structure of a monoamine transporter from the SLC6 family has recently been proposed to have binding sites not only for the monoamine, but also for two sodium ions (Figure 2-2A). In addition, these transporters may exist as dimers, or two copies working together with each other, but the manner in which they cooperate is not yet well understood and is not shown in the figures. There are other sites on this transporter – not well defined – for drugs such as antidepressants, which bind to the transporter and inhibit reuptake of monoamines but do not bind to the substrate site and are not transported into the neuron (thus they are allosteric, meaning “other site”).
In the absence of sodium, there is low affinity of the monoamine transporter for its monoamine substrate, and thus binding of neither sodium nor monoamine. An example of this is shown for the serotonin transporter SERT in Figure 2-2A, where some of the transport “wagons” have flat tires, indicating no binding of sodium as well as absence of binding of serotonin to its substrate binding site, since the transporter has low affinity for serotonin in the absence of sodium. The allosteric site for antidepressant binding is also empty (the front seat in Figure 2-2A). However, in the presence of sodium ions, the tires are “inflated” by sodium binding and serotonin can also bind to its substrate site on SERT. The situation is now primed for serotonin transport back into the serotonergic neuron, along with cotransport of sodium and chloride down the gradient and into the neuron and countertransport of potassium out of the neuron (Figure 2-2A). But if a drug binds to an inhibitory allosteric site on SERT, this reduces the affinity of the serotonin transporter SERT for its substrate serotonin, and serotonin binding is prevented.
Why does this matter? Blocking the presynaptic monoamine transporter has a huge impact on neurotransmission at any synapse that utilizes that neurotransmitter. The normal recapture of neurotransmitter by the presynaptic neurotransmitter transporter in Figure 2-2A keeps the levels of this neurotransmitter from accumulating in the synapse. Normally, following release from the presynaptic neuron, neurotransmitters only have time for a brief dance on their synaptic receptors, and the party is soon over because the monoamines climb back into the presynaptic neuron on their transporters (Figure 2-2A). If one wants to enhance normal synaptic activity of these neurotransmitters, or restore their diminished synaptic activity, this can be accomplished by blocking these transporters. Although this might not seem to be a very dramatic thing, the fact is that this alteration in chemical neurotransmission – namely the enhancement of synaptic monoamine action – is thought to underlie the clinical effects of all the agents that block monoamine transporters, including most known antidepressants and stimulants. Specifically, many antidepressants enhance serotonin, norepinephrine, or both, due to actions on SERT and/or NET. Some antidepressants act on DAT, as do stimulants. Also, recall that many antidepressants that block monoamine transporters are also effective anxiolytics, reduce neuropathic pain, and have additional therapeutic actions as well. Thus, it may come as no surprise that drugs that block monoamine transporters are among the most frequently prescribed psychotropic drugs. In fact, about a third of the currently prescribed essential psychotropic drugs act by targeting one or more of the three monoamine transporters.
Other neurotransmitter transporters (SLC6 and SLC1 gene families) as targets of psychotropic drugs
In addition to the three transporters for monoamines discussed in detail above, there are several other transporters for various different neurotransmitters or their precursors. Although this includes a dozen additional transporters, there is only one psychotropic drug used clinically that is known to bind to any of these transporters. Thus, there is a presynaptic transporter for choline, the precursor to the neurotransmitter acetylcholine, but no known drugs target this transporter. There are also several transporters for the ubiquitous inhibitory neurotransmitter GABA, known as GAT1–4 (Table 2-2). Although debate continues about the exact localization of these subtypes at presynaptic neurons, neighboring glia, or even postsynaptic neurons, it is clear that a key presynaptic transporter of GABA is the GAT1 transporter, which is selectively blocked by the anticonvulsant tiagabine, thereby increasing synaptic GABA concentrations. In addition to anticonvulsant actions, this increase in synaptic GABA may have therapeutic actions in anxiety, sleep disorders, and pain. No other inhibitors of this transporter are available for clinical use.
Finally, there are multiple transporters for two amino acid neurotransmitters, glycine and glutamate (Table 2-2). There are no drugs utilized in clinical practice that are known to block glycine transporters, although new agents are in clinical trials for treating schizophrenia. The glycine transporters, along with the choline and GABA transporters, are all members of the same family to which the monoamine transporters belong and have a similar structure (Figure 2-2A, Tables 2-1 and 2-2). However, the glutamate transporters belong to a unique family, SLC1, and have a unique structure and somewhat different functions compared to those transporters of the SLC6 family (Table 2-2).
Specifically, there are several transporters for glutamate, known as excitatory amino acid transporters 1–5, or EAAT1–5 (Table 2-2). The exact localization of these various transporters at presynaptic neurons, postsynaptic neurons, or glia is still under investigation, but the uptake of glutamate into glia is well known to be a key system for recapturing glutamate for reuse once it has been released. Transport into glia results in conversion of glutamate into glutamine, and then glutamine enters the presynaptic neuron for reconversion back into glutamate. No drugs utilized in clinical practice are known to block glutamate transporters.
One difference between transport of neurotransmitters by the SLC6 gene family and transport of glutamate by the SLC1 gene family is that glutamate does not seem to cotransport chloride with sodium when it also cotransports glutamate. Also, glutamate transport is almost always characterized by the countertransport of potassium, whereas this is not always the case with SLC6 gene family transporters. Glutamate transporters may work together as trimers rather than dimers, as the SLC6 transporters seem to do. The functional significance of these differences remains obscure, but may become more apparent if clinically useful psychopharmacologic agents that target glutamate transporters are discovered. Since it may often be desirable to diminish rather than enhance glutamate neurotransmission, the future utility of glutamate transporters as therapeutic targets is also unclear.
Where are the transporters for histamine and neuropeptides?
It is an interesting observation that apparently not all neurotransmitters are regulated by reuptake transporters. The central neurotransmitter histamine apparently does not have a presynaptic transporter (although it is transported into synaptic vesicles by VMAT2, the same transporter used by the monoamines – see Figure 2-2B). Histamine’s inactivation is thus thought to be entirely enzymatic. The same can be said for neuropeptides, since reuptake pumps and presynaptic transporters have not been found for them, and are thus thought to be lacking for this class of neurotransmitter. Inactivation of neuropeptides is apparently by diffusion, sequestration, and enzymatic destruction, but not by presynaptic transport. It is always possible that a transporter will be discovered in the future for some of these neurotransmitters, but at the present time there are no known presynaptic transporters for either histamine or neuropeptides.
Vesicular transporters: subtypes and function
Vesicular transporters for the monoamines (VMATs) are members of the SLC18 gene family and have already been discussed above. They are shown in Figure 2-2B and listed in Table 2-3, as is the vesicular transporter for acetylcholine – also a member of the SLC18 gene family but known as VAChT. The GABA vesicular transporter is a member of the SLC32 gene family and is called VIAAT (vesicular inhibitory amino acid transporter; Figure 2-2B and Table 2-3). Finally, vesicular transporters for glutamate, called vGluT1–3 (vesicular glutamate transporters 1, 2, and 3) are members of the SLC17 gene family – and are also shown in Figure 2-2B and listed in Table 2-3. The SV2A transporter is a novel twelve-transmembrane-region synaptic vesicle transporter of uncertain mechanism and with unclear substrates; it is localized within the synaptic vesicle membrane and binds the anticonvulsant levetiracetam, perhaps interfering with neurotransmitter release and thereby reducing seizures.
How do neurotransmitters get inside synaptic vesicles? In the case of vesicular transporters, storage of neurotransmitters is facilitated by a proton ATPase, known as the “proton pump,” that utilizes energy to pump positively charged protons continuously out of the synaptic vesicle (Figure 2-2B). The neurotransmitters can then be concentrated against a gradient by substituting their own positive charge inside the vesicle for the positive charge of the proton being pumped out. Thus, neurotransmitters are not so much transported as “antiported” – i.e., they go in while the protons are actively transported out, keeping charge inside the vesicle constant. This concept is shown in Figure 2-2B for the VMAT transporting dopamine in exchange for protons. Contrast this with Figure 2-2A, where a monoamine transporter on the presynaptic membrane is cotransporting a monoamine along with sodium and chloride, but with the help of a sodium-potassium ATPase (sodium pump) rather than a proton pump.
Vesicular transporters (SLC18 gene family) as targets of psychotropic drugs
Vesicular transporters for acetylcholine (SLC18 gene family), GABA (SLC32 gene family), and glutamate (SLC17 gene family) are not known to be targeted by any drug utilized by humans. However, vesicular transporters for monoamines in the SLC18 gene family, or VMATs, particularly those in dopamine and norepinephrine neurons, are potently targeted by several drugs including amphetamine, tetrabenazine, and reserpine. Amphetamine thus has two targets: monoamine transporters as well as VMATs. In contrast, other stimulants such as methylphenidate and cocaine target only the monoamine transporters, and in much the same manner as described for antidepressants (see Chapter 7).
G-protein-linked receptors
Structure and function
Another major target of psychotropic drugs is the class of receptors linked to G proteins. These receptors all have the structure of seven transmembrane regions, meaning that they span the membrane seven times (Figure 2-1). Each of the transmembrane regions clusters around a central core that contains a binding site for a neurotransmitter. Drugs can interact at this neurotransmitter binding site or at other sites (allosteric sites) on the receptor. This can lead to a wide range of modifications of receptor actions due to mimicking or blocking, partially or fully, the neurotransmitter function that normally occurs at this receptor. These drug actions can thus change downstream molecular events such as which phosphoproteins are activated or inactivated and therefore which enzymes, receptors, or ion channels are modified by neurotransmission. Such drug actions can also change which genes are expressed, and thus which proteins are synthesized and which functions are amplified, from synaptogenesis, to receptor and enzyme synthesis, to communication with downstream neurons innervated by the neuron with the G-protein-linked receptor.
These actions on neurotransmission by G-protein-linked receptors are described in detail in Chapter 1 on signal transduction and chemical neurotransmission. The reader should have a good command of the function of G-protein-linked receptors and their role in signal transduction from specific neurotransmitters, as described in Chapter 1, in order to understand how drugs acting at G-protein-linked receptors modify the signal transduction that arises from these receptors. This is important to understand because such drug-induced modifications in signal transduction from G-protein-linked receptors can have profound actions on psychiatric symptoms. In fact, the single most common action of psychotropic drugs utilized in clinical practice is to modify the actions of G-protein-linked receptors, resulting in either therapeutic actions or side effects. Here we will describe how various drugs stimulate or block these receptors, and throughout the textbook we will show how specific drugs acting at specific G-protein-linked receptors have specific actions on specific psychiatric disorders.
G-protein-linked receptors as targets of psychotropic drugs
G-protein-linked receptors are a large superfamily of receptors that interact with many neurotransmitters and with many psychotropic drugs (Figure 2-1B). There are numerous ways to subtype these receptors, but pharmacologic subtypes are perhaps the most important to understand for clinicians who wish to target specific receptors with psychotropic drugs utilized in clinical practice. That is, the natural neurotransmitter interacts at all of its receptor subtypes, but many drugs are more selective than the neurotransmitter itself for certain receptor subtypes and thus define a pharmacologic subtype of receptor at which they specifically interact. This is not unlike the concept of the neurotransmitter being a master key that opens all the doors, and a drug that interacts at pharmacologically specific receptor subtypes functioning as a specific key opening only one door. Here we will develop the concept that drugs have many different ways of interacting at pharmacologic subtypes of G-protein-linked receptors, which occur across an agonist spectrum (Figure 2-3).
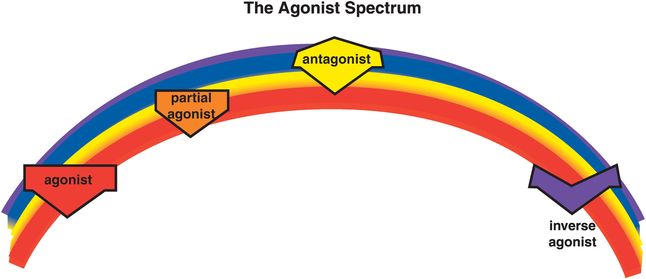
Figure 2-3. Agonist spectrum. Shown here is the agonist spectrum. Naturally occurring neurotransmitters stimulate receptors and are thus agonists. Some drugs also stimulate receptors and are therefore agonists as well. It is possible for drugs to stimulate receptors to a lesser degree than the natural neurotransmitter; these are called partial agonists or stabilizers. It is a common misconception that antagonists are the opposite of agonists because they block the actions of agonists. However, although antagonists prevent the actions of agonists, they have no activity of their own in the absence of the agonist. For this reason, antagonists are sometimes called “silent.” Inverse agonists, on the other hand, do have opposite actions compared to agonists. That is, they not only block agonists but can also reduce activity below the baseline level when no agonist is present. Thus, the agonist spectrum reaches from full agonists to partial agonists through to “silent” antagonists and finally inverse agonists.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


