Transanal Pull Through for Hirschsprung Disease
Jacob C. Langer
Introduction
Hirschsprung disease is characterized by absence of ganglion cells within the submucosal and myenteric plexuses in the distal bowel, most commonly involving the rectum and part of the sigmoid colon. Diagnosis is suspected on clinical grounds in newborns and infants with distal intestinal obstruction or in older children with severe chronic constipation. Contrast enema usually shows a transition zone between the normal and the aganglionic bowel (Fig. 1), although the contrast enema may be falsely negative in up to 10% of neonates with Hirschsprung disease. The diagnosis is confirmed based on a rectal biopsy showing aganglionosis, usually associated with histological evidence of hypertrophic nerves and abnormal cholinesterase and calretinin staining. In neonates this is done using a suction device, and in older children a deeper biopsy must be done. In all cases, care must be taken to biopsy at least 1 to 2 cm above the dentate line, since there is a normal hypoganglionic region below that level.
The treatment of Hirschsprung disease is surgical, and the principle of the operation is to remove the aganglionic colon and anastomose normally innervated bowel to the anus, while preserving anal sphincter function. The first such operation, described by Swenson in the late 1940s, involved complete rectal resection and primary end-to-end anastomosis just above the anal sphincter. Two additional operations were subsequently described and popularized. The Soave procedure involves a submucosal dissection with an endorectal pull through, leaving a rectal muscular cuff, and the Duhamel operation brings the normal bowel through the avascular plane behind the aganglionic rectum, with a stapled anastomosis joining the two lumens.
Because these children often presented with severe malnutrition or enterocolitis, a preliminary colostomy was usually done, followed by a pull-through procedure many months later. Earlier recognition and diagnosis of the disease led a number of surgeons in the 1980s to report single-stage pull-through procedures even in small infants. Over the past few decades, one-stage operations have become increasingly popular and are considered to be safe, cost-effective, and the approach of choice for most children with Hirschsprung disease. A stoma is still considered to be appropriate in children with severe enterocolitis, very dilated proximal bowel, total colonic aganglionosis, or lack of adequate pathology support.
In the early 1990s, Georgeson described a minimal access approach, consisting of a laparoscopic biopsy to identify the transition zone, laparoscopic mobilization of the rectum below the peritoneal reflection, a short endorectal mucosal dissection from below, and an anastomosis from below after prolapsing and excising the rectum. Laparoscopic approaches were subsequently described for the Duhamel and Swenson operations. The transanal Soave procedure represented a natural evolution from the laparoscopic operation, and initial series in children with Hirschsprung disease were published by de la Torre and Langer in the late 1990s. The transanal approach has the principal benefit of avoiding the need for intra-abdominal mobilization of the rectum via either laparotomy or laparoscopy. Both the laparoscopic and the transanal approaches are associated with less pain, shorter hospital stay, and a better cosmetic result than open surgery, but there have not been any studies comparing the laparoscopic to the transanal approach. However, the transanal pull through can be done by any pediatric surgeon, including those without laparoscopic skills, and by pediatric surgeons in parts of the world where access to appropriately miniaturized laparoscopic equipment is limited.
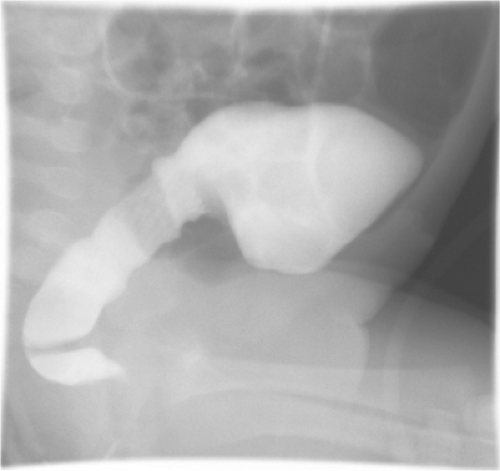 Fig. 1. Contrast enema demonstrating a typical transition zone between normal and aganglionic bowel at the rectosigmoid junction. The lateral view is the most effective for seeing the transition zone. |
Infants with intestinal obstruction or enterocolitis should undergo rapid resuscitation with intravenous fluids, antibiotics, and a nasogastric tube. Early decompression of the colon using digital rectal stimulation and/or irrigations through a rectal tube is important to prevent and treat enterocolitis and to decrease the diameter of the colon. Children with associated abnormalities such as cardiac disease or congenital central hypoventilation syndrome must have these problems investigated and managed prior to definitive surgical repair. Once the child has been stabilized, surgery can be done semi-electively. While waiting for surgery most children can be fed breast milk or an elemental formula, in combination with rectal stimulations or irrigations, although some will require ongoing nasogastric decompression and parenteral nutrition.
The older child who presents with severe constipation and an extremely dilated colon may require weeks or months of irrigations to bring the colon to a more normal size
prior to definitive surgery. Some children with particularly dilated or thickened proximal colon may require a colostomy to achieve adequate decompression.
prior to definitive surgery. Some children with particularly dilated or thickened proximal colon may require a colostomy to achieve adequate decompression.
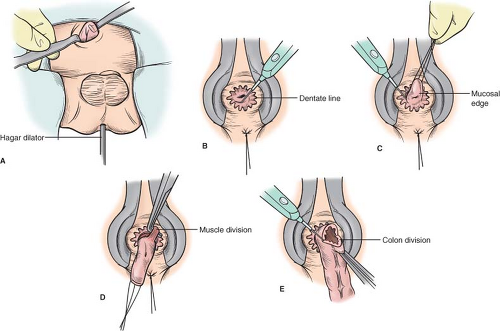 Fig. 2. A: A Hagar dilator is passed through the anus to push the sigmoid colon up toward the incision, and the colon is biopsied until normal ganglion cells are identified on frozen section. B: The dentate line is clearly visualized and a mucosal incision is made 0.5 cm above the dentate line. C: Fine silk sutures are placed circumferentially along the mucosal edge and the mucosa is dissected off the underlying muscle for ∼2 cm. D: The muscle is divided and the dissection is carried proximally on the rectal wall until the site of the normal biopsy is reached. E: The colon is divided and the anastomosis performed. |
There are three important anatomic structures or planes that must be recognized during the safe and correct performance of a transanal pull through. Firstly, the anorectal squamocolumnar junction (dentate or pectinate line) must be clearly identified, and the initial mucosal incision must be made high enough that the transitional epithelium is preserved. If this epithelium is damaged, sensation will be abnormal and continence will be impaired. Secondly, the relatively bloodless submucosal plane of the rectum permits the surgeon to avoid injury to the vagina or prostate during the initial dissection. Thirdly, the plane on the serosal surface of the rectum is also relatively bloodless, and the principle of staying in this plane will prevent injury to pelvic structures.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


