Treatment at the Level of the Protein
In many situations, if a mutant protein product is made, it may be possible to increase its function. For example, the stability or function of a mutant protein with some residual function may be further increased. With enzymopathies, the improvement in function obtained by this approach is usually very small, on the order of a few percent, but this increment is often all that is required to restore biochemical homeostasis.
Enhancement of Mutant Protein Function with Small Molecule Therapy
Small molecules are compounds with molecular weights in the few hundreds to thousands. They include vitamins, nonpeptide hormones, and indeed most drugs, whether synthesized by organic chemists or isolated from nature. A new strategy for identifying potential drugs is to use high-throughput screening of chemical compound libraries, often containing tens of thousands of known chemicals, against a drug target, such as the protein whose function is disrupted by a mutation. As we will discuss, two drugs that are now FDA approved for the treatment of some patients with CF, and another that is investigational, were discovered using such high-throughput screens. Progress in the development of these drugs represents a new frontier with great potential for the treatment of genetic disease.
Small Molecule Therapy to Allow Skipping over Nonsense Codons.
Nonsense mutations account for 11% of defects in the human genome. Approximately 9% of all CFTR alleles are nonsense mutations, and approximately 50% of Ashkenazi Jewish patients with CF carry at least one CFTR allele with a premature stop codon (e.g., Arg553Stop). A potentially ideal therapeutic approach (other than gene therapy) for patients with a nonsense mutation would be a safe drug that encourages the translational apparatus to misread the stop codon by a transfer RNA (tRNA) that is near-cognate to the stop codon tRNA. If the amino acid thereby inserted into the polypeptide by that tRNA still produces a functional protein, the activity of the protein would be restored. An event of this type, for example, would convert the CFTR Arg553Stop mutation to 553Tyr, a substitution that generates a CFTR peptide with nearly normal properties. High-throughput chemical screens for a drug of this type identified ataluren (PTC124), and evidence suggests that it is most effective in allowing read-through of TGA nonsense codons. Moreover, studies in model organisms have firmly demonstrated that it can correct the mutant phenotype of some nonsense mutations. Ataluren has not been established to be clinically effective, but a Phase III clinical trial in CF patients carrying at least one nonsense mutation showed a promising trend toward statistically significant improvement in lung function, and a follow-up trial is underway. Even if ataluren proves ineffective in humans, thousands of other small molecules are being examined in laboratories around the world to identify novel nontoxic compounds that facilitate the skipping of nonsense codons, not only for the treatment of CF but also for Duchenne muscular dystrophy patients carrying nonsense codons, as well as other diseases. Safe, effective drugs of this type will have a major impact on the treatment of inherited disease.
Small Molecules to Correct the Folding of Mutant Membrane Proteins: Pharmacological Chaperones.
Some mutations in membrane proteins may disrupt their ability to fold, pass through the endoplasmic reticulum, and be trafficked to the plasma membrane. These mutant proteins are recognized by the cellular protein quality control machinery, trapped in the endoplasmic reticulum, and prematurely degraded by the proteosome. The ΔF508 deletion of the CFTR protein—which constitutes 65% of all CF mutations worldwide—is perhaps the best-known example (see Fig. 12-15) of a mutation that impairs trafficking of a membrane protein. If the folding/trafficking defect could be overcome to increase the abundance of CFTR channels at the apical surface of the cell by 20% to 25%, it is thought that a clinical benefit would be obtained, because once the ΔF508 CFTR protein reaches the cell surface, it is an effective Cl− channel.
Small molecule screens to identify compounds that can serve as a chaperone to prevent misfolding and correct the ΔF508 CFTR trafficking defect in in vitro assay systems have identified lumacaftor (VX-809) as an effective, although incomplete, corrector of this specific CFTR mutant polypeptide (see Fig. 13-7). Lumacaftor interacts directly with the mutant CFTR to stabilize its three-dimensional structure, specifically correcting the underlying trafficking defect and enhancing Cl− transport. Although monotherapy with lumacaftor had no clinical benefits, a recently completed Phase III clinical trial using lumacaftor together with another small molecule, ivacaftor (VX-770), discussed later, showed significant improvements in lung function in homozygous ΔF508 CFTR patients. This finding is notable because it is the first treatment shown to have a favorable impact on the primary biochemical defect in patients carrying the most common CFTR allele, ΔF508. Ongoing studies of the long-term effectiveness and safety of the lumacaftor-ivacaftor combination therapy are in progress. Irrespective of their success, this example is a milestone in medical genetics, because it establishes the principle that molecular chaperones can have clinical benefits in the treatment of monogenic disease.
Small Molecules to Increase the Function of Correctly Trafficked Mutant Membrane Proteins.
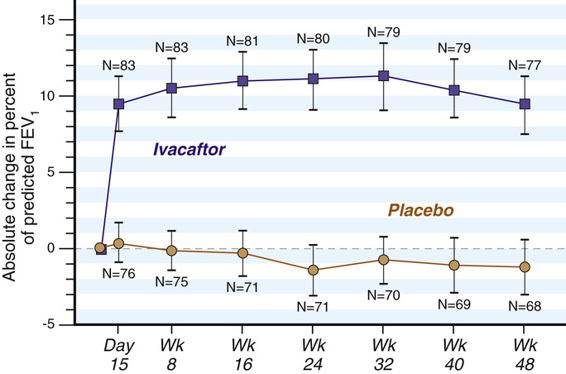
Amino acid substitutions in membrane proteins may not disrupt the trafficking of the mutant polypeptide to the plasma membrane, but rather interfere with its function at the cell surface. Small molecule screens for new treatments for CF have also led this area of drug discovery. Screens for so-called potentiators—molecules that could enhance the function of mutant CFTR proteins that are correctly positioned at the cell surface—identified ivacaftor (VX-770), which improves the Cl− transport of some mutant CFTR proteins, such as the Gly551Asp CFTR missense mutation (see Fig. 12-15) that inactivates anion transport; this allele is carried by 4% to 5% of all CF patients. In one clinical trial, patients carrying at least one Gly551Asp allele experienced a significant improvement in lung function (Fig. 13-8), weight gain, respiratory symptoms, and a decline in sweat Cl−. Ivacaftor is presently FDA approved for the treatment of eight other CFTR missense mutations, and more alleles will certainly be added to this group. Although fewer than 200 CF patients in the United States have one of these eight alleles, the allele-specific indications for ivacaftor treatment highlight both the benefits and dilemmas of personalized medicine for genetic disease: effective drugs can be discovered, but they may be effective only in a relatively small numbers of individuals. Moreover, at present ivacaftor is extremely expensive, costing approximately $300,000 per year.
Small Molecules to Enhance the Function of Mutant Enzymes: Vitamin-Responsive Inborn Errors of Metabolism.
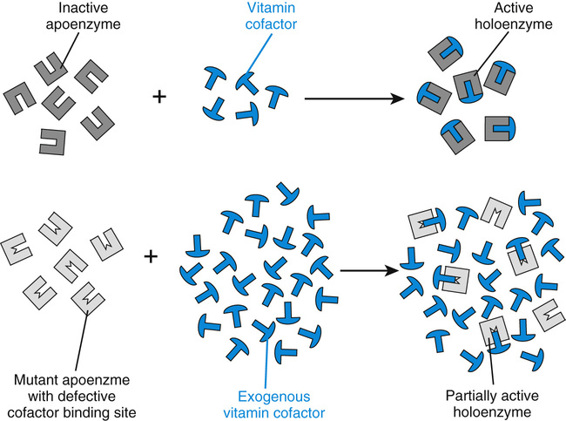
The biochemical abnormalities of a number of inherited metabolic diseases may respond, sometimes dramatically, to the administration of large amounts of the vitamin cofactor of the enzyme impaired by the mutation (Table 13-3). In fact, the vitamin-responsive inborn errors are among the most successfully treated of all genetic diseases. The vitamins used are remarkably nontoxic, generally allowing the safe administration of amounts 100 to 500 times greater than those required for normal nutrition. In homocystinuria due to cystathionine synthase deficiency (see Fig. 12-8), for example, approximately 50% of patients respond to the administration of high doses of pyridoxine (vitamin B6, the precursor of pyridoxal phosphate, the cofactor for the enzyme), an example—as we saw earlier in the case of BH4 administration in PKU—of cofactor responsiveness in a metabolic disease. In most of these responsive patients, homocystine completely disappears from the plasma, even though the increase in hepatic cystathionine synthase activity is usually only a fewfold, from 1.5% to 4.5% of control activity. The increased pyridoxal phosphate concentrations may stabilize the mutant enzyme or overcome reduced affinity of the mutant enzyme for the cofactor (Fig. 13-9). In any case, vitamin B6 treatment substantially improves the clinical course of the disease in responsive patients. Nonresponsive patients generally carry null alleles and therefore have no residual cystathionine synthase activity to augment.
TABLE 13-3
Treatment of Genetic Disease at the Level of the Mutant Protein
| Strategy | Example | Status |
| Enhancement of Mutant Protein Function | ||
| Small molecules that facilitate translational “skipping” over mutant stop codons | Ataluren in the 10% of cystic fibrosis patients with nonsense mutations in the CFTR gene | Investigational in CF: confirmatory Phase III clinical trial was begun in 2014 |
| Small molecule “correctors” that increase the trafficking of the mutant protein through the ER to the plasma membrane | Lumacaftor (VX-809) to increase the abundance of the ΔF508 mutant CFTR protein at the apical membrane of epithelial cells in CF patients | Investigational: very promising improvements in lung function in ΔF508 homozygotes, when used in combination with ivacaftor; expensive |
| Small molecule “potentiators” that increase the function at the cell membrane of correctly trafficked membrane proteins | Ivacaftor (VX-770) used alone to enhance the function of specific mutant CFTR proteins at the epithelial apical membrane | FDA approved for the treatment of CF patients carrying specific alleles; expensive |
| Vitamin cofactor administration to increase the residual activity of the mutant enzyme | Vitamin B6 for pyridoxine-responsive homocystinuria | Treatment of choice in the 50% of cystathionine synthase patients who are responsive |
| Protein Augmentation | ||
| Replacement of an extracellular protein | Factor VIII in hemophilia A | Well-established, effective, safe |
| Extracellular replacement of an intracellular protein | Polyethylene glycol–modified adenosine deaminase (PEG-ADA) in ADA deficiency | Well-established, safe, and effective, but costly; now used principally to stabilize patients before gene therapy or HLA-matched bone marrow transplantation |
| Replacement of an intracellular protein—cell targeting | β-glucocerebrosidase in non-neuronal Gaucher disease | Established; biochemically and clinically effective; expensive |
Protein Augmentation
The principal types of protein augmentation are summarized in Table 13-3. Protein augmentation is a routine therapeutic approach in only a few diseases, all involving proteins whose principal site of action is in the plasma or extracellular fluid. The prime example is the prevention or arrest of bleeding episodes in patients with hemophilia (Case 21) by the infusion of plasma fractions enriched for the appropriate factor. The decades of experience with this disease illustrate the problems that can be anticipated as new strategies for replacing other, particularly intracellular, polypeptides are attempted. These problems include the difficulty and cost of procuring sufficient amounts of the protein to treat all patients at the optimal frequency, the need to administer the protein at a frequency consistent with its half-life (only 8 to 10 hours for factor VIII), and the formation of neutralizing antibodies in some patients (5% of classic hemophiliacs).
Enzyme Replacement Therapy: Extracellular Administration of an Intracellular Enzyme
Adenosine Deaminase Deficiency.
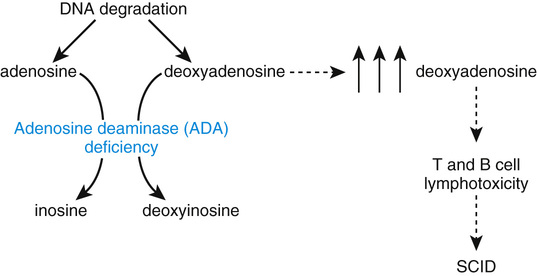
Adenosine deaminase (ADA) is a critical enzyme of purine metabolism that catalyzes the deamination of adenosine to inosine and of deoxyadenosine to deoxyinosine (Fig. 13-10). The pathology of ADA deficiency, an autosomal recessive disease, results entirely from the accumulation of toxic purines, particularly deoxyadenosine, in lymphocytes. A profound failure of both cell-mediated (T-cell) and humoral (B-cell) immunity results, making ADA deficiency one cause of severe combined immunodeficiency (SCID). Untreated patients die of infection within the first 2 years of life. The long-term treatment of ADA deficiency is rapidly evolving, with gene therapy (see later section) now a strong alternative to bone marrow transplantation from a fully human leukocyte antigen (HLA) compatible donor. The administration of a modified form of the bovine ADA enzyme, described in the next section, is no longer a first choice for long-term management, but it is an effective stabilizing measure in the short term until these other treatments can be used.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


