analysis to be calcium oxalate) is commonly found in the colloid of normal or diseased thyroids (20). Small collections of C cells are situated within the confines of the basement membrane of the thyroid follicles. Difficult to identify by ordinary histologic stains, the normal C cells can be identified by immunostaining for calcitonin (21).
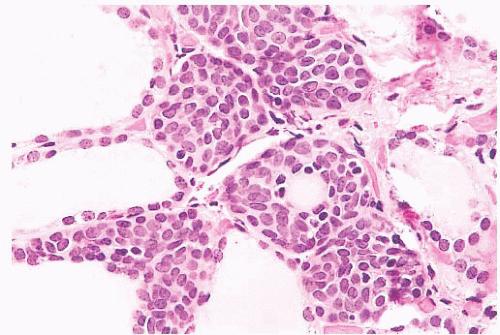 FIGURE 13.1 Ultimobranchial body rest in thyroid showing solid cell nests with focal cyst formation. The ultimobranchial body is believed to give rise to the thyroid C cells. |
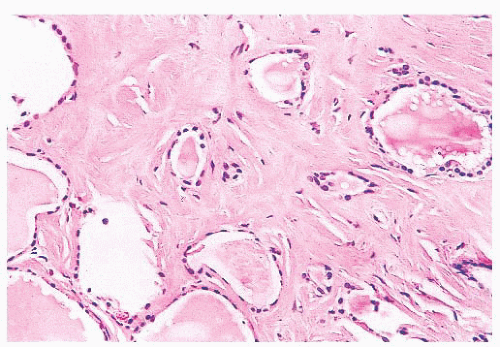 FIGURE 13.2 High-power view of lingual thyroid showing thyroid follicle surrounded by dense fibrous tissue. This was an incidental finding at surgery in a patient with head and neck squamous cell carcinoma. |
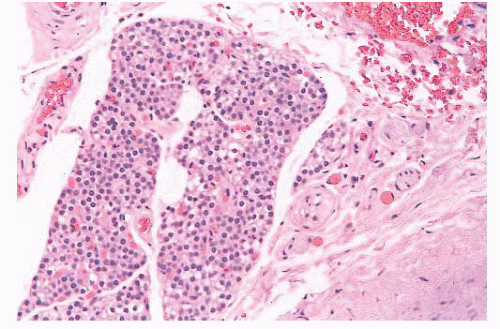 FIGURE 13.3 Parathyroid tissue involving capsule of the thyroid. Most parathyroid tissue associated with the thyroid is present within the thyroid capsule. Occasionally intrathyroidal parathyroid tissue may be seen. |
purulence. Microscopically, acute inflammation with microabscess formation is present. Microorganisms may be seen. A variety of organisms cause thyroiditis including bacteria, fungi, and viruses (26,27). In individuals with human immunodeficiency virus infection, the occurrence of Pneumocystis jiroveci infection has been reported.
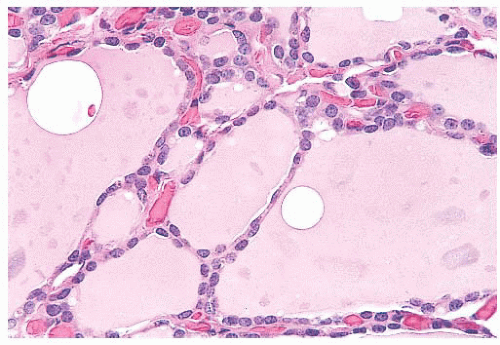 FIGURE 13.4 Normal thyroid follicles lined by a single layer of flat cuboidal follicular epithelial cells. A rich vascular network is observed between the follicles. |
TABLE 13.1 Key Features of Thyroiditides | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
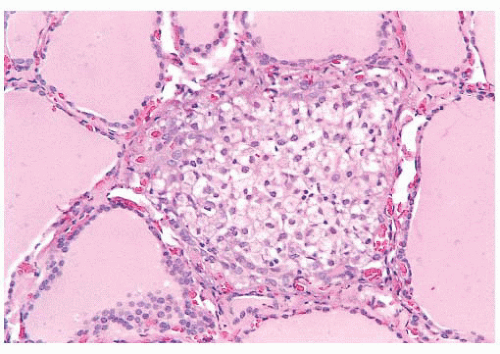 FIGURE 13.5 “Palpation thyroiditis” in a thyroid removed for papillary thyroid carcinoma. This form of thyroiditis is characterized by the presence of histiocytes and lymphocytes, is usually focal, and is associated with minor trauma to the gland. |
lymphoma, B-cell type (46,47). In addition, patients with Hashimoto disease may be prone to the development of plasmacytomas within the gland (48). A peculiar variant of mucoepidermoid carcinoma, known as sclerosing mucoepidermoid carcinoma with eosinophilia, has been recognized in patients with Hashimoto disease (49). Whether there is an increase in the incidence of papillary carcinoma—especially microcarcinoma— is still debated.
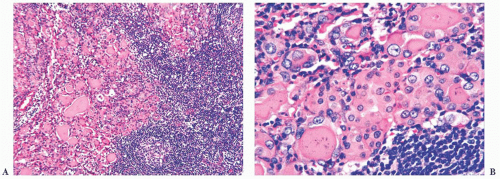 FIGURE 13.6 (A) The thyroid in Hashimoto disease shows a dense lymphoplasmacytic infiltrate with germinal center formation. The follicular epithelium often shows Hürthle cell change. (B) Oftentimes, the follicular epithelium adjacent to the inflammatory cell infiltrate of autoimmune thyroiditis shows “clear” chromatin similar to the nuclear changes seen in papillary thyroid carcinoma. These changes are often seen scattered throughout the inflamed gland and should not be misinterpreted as papillary carcinoma. |
Chronic thyroiditis, oxyphilic: This group contains patients with classic Hashimoto disease histology.
Chronic thyroiditis, mixed: This group shows less of an infiltrate than group 1 with minimal fibrosis. Patients demonstrate normal thyroid, hyperthyroidism, or hypothyroidism.
Chronic thyroiditis, hyperplastic: This group shows glandular hyperplasia associated with only a small lymphocytic reaction. Most patients are hyperthyroid.
Chronic thyroiditis, focal: This group shows only a focal lymphocytic reaction, and most patients are euthyroid.
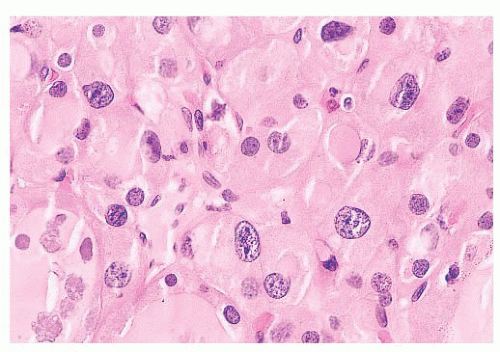 FIGURE 13.7 This nodule from a patient with nodular goiter shows random nuclear atypia. This change is often seen in the thyroid after radiation exposure. |
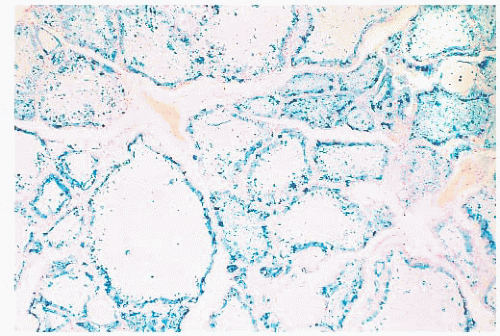 FIGURE 13.8 A Prussian blue stain for iron in a patient with hemochromatosis shows extensive iron deposition in follicular epithelial cells. |
of fibrovascular (occasionally just fibrous) tissue lined by one or occasionally several layers of cells with crowded oval nuclei (Fig. 13.10). In contrast, hyperplasia of thyroid follicles may sometimes exaggerate into papillary change; there is infolding of the lining epithelium composed of columnar cells with basal round and uniform nuclei. There is either no central core or a core of edematous or myxomatous paucicellular stroma often including small follicles (subfollicle formation) (71,110,124).
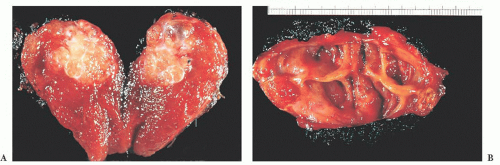 FIGURE 13.9 (A) Gross picture of a papillary thyroid carcinoma showing a yellow-white infiltrative mass with evidence of fibrous strands. (B) Gross picture of a papillary thyroid carcinoma with extensive cyst formation. Note the smooth lining to the cyst wall. This lesion was also associated with cystic lymph node metastases, which were the first presenting sign of tumor. These lesions can be difficult to diagnose, particularly on fineneedle aspiration, because the papillary carcinoma cells may be only focally present in the cyst wall. |
TABLE 13.2 Key Histopathologic Features of Commonly Encountered Variants of Papillary Thyroid Carcinoma | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
are often found. Another characteristic of the papillary cancer nucleus is the nuclear groove (97,129). Nuclear grooves may be seen in other thyroid lesions including Hashimoto disease, adenomatous hyperplasia, and diffuse hyperplasia as well as in follicular adenomas (particularly hyalinizing trabecular neoplasm) (130,131). For the most part, nuclear grooves are more commonly seen in papillary carcinoma than in other thyroid lesions; however, the mere presence of nuclear grooves is not diagnostic for papillary carcinoma.
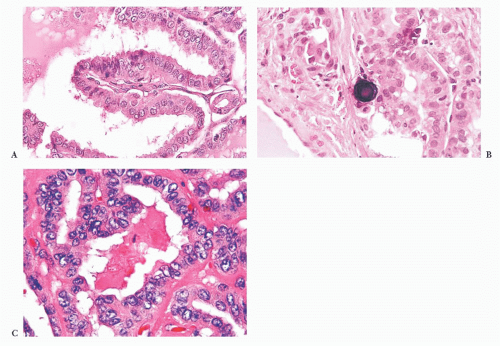 FIGURE 13.10 (A) Microscopic examination of a papilla in papillary carcinoma. Even at this power, nuclear clearing, elongation, and overlapping are identified. The papillary group shows a nonedematous vascular core (200×). (B) Psammoma body formation is common in papillary thyroid carcinoma. (C) High-power view of papillary carcinoma nuclei showing nuclear clearing, enlargement, overlapping, and intranuclear grooves (400×). |
(153,154); however, multivariate analysis has not shown that ploidy is an independent prognostic factor.
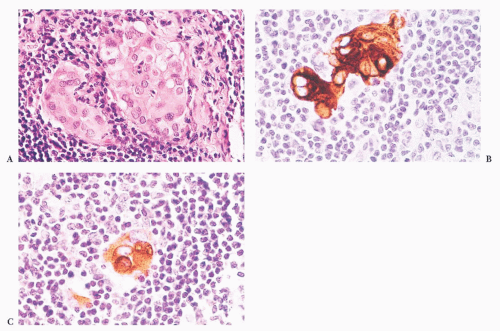 FIGURE 13.11 (A) Small focus of epithelial cells identified in a neck dissection for head and neck squamous carcinoma. (B) Immunostaining for cytokeratin reveals that these cells are epithelial in nature. (C) Immunostaining for thyroglobulin confirms that the epithelial cells are consistent with metastatic thyroid carcinoma. A subsequent thyroidectomy confirmed the presence of papillary carcinoma. |
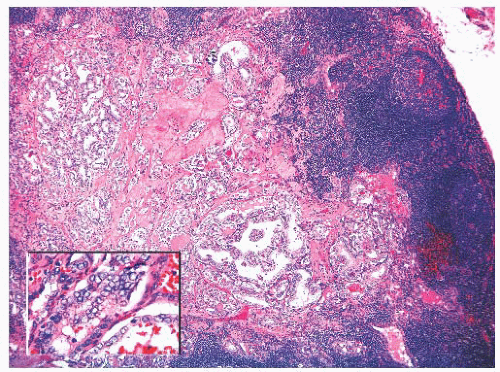 FIGURE 13.12 Metastatic papillary carcinoma in a neck lymph node. The inset shows the diagnostic nuclear features. |
of fusion of tyrosine kinase domain of RET to the 5’ portion of the various genes (155). To date, 11 novel types of rearrangements have been described in papillary carcinoma. RET/PTC1 and RET/PTC3 are the most common forms that occur in sporadic papillary carcinoma. RET/PTC1 is formed by fusion of RET to H4, and RET/PTC3 occurs as a result of fusion of RET to the ELE1 gene (159).
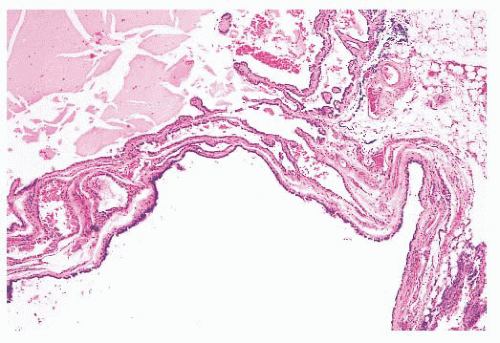 FIGURE 13.13 Metastatic papillary carcinoma with extensive cyst formation in a neck lymph node. These lesions may be confused clinically with branchial cleft cysts, particularly in patients with no known history of thyroid carcinoma. |
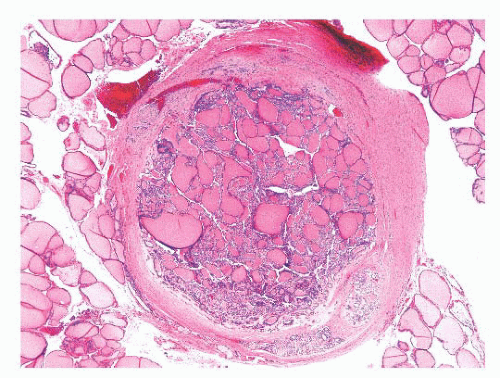 FIGURE 13.14 Papillary microcarcinoma in a patient with a benign follicular nodule. This lesion was an incidental finding in the thyroid lobectomy specimen. |
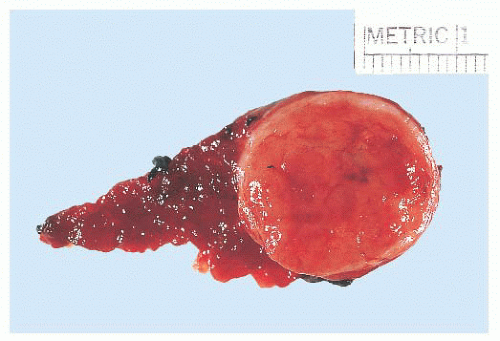 FIGURE 13.15 The follicular variant of papillary carcinoma may grossly appear as a circumscribed nodule resembling a follicular adenoma. Microscopic examination shows a follicular architecture with nuclear features of typical papillary thyroid carcinoma. |
PAX8/peroxisome proliferator-activated receptor-γ (PPAR-γ) rearrangements, thus resembling more true follicular carcinoma than papillary subtype. It is unusual to find BRAF mutations in encapsulated follicular variant of papillary carcinoma (158,213,214). It has been shown that FVPTC demonstrates a distinct BRAF (K601E) mutation from the usual BRAF (V600E) mutation found in classic and tall cell variant of PTC (212,213,215).
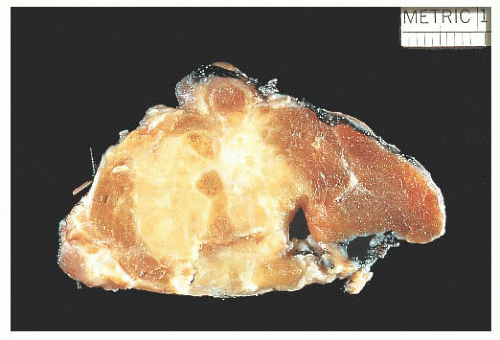 FIGURE 13.16 Gross photo of a tall cell variant of papillary carcinoma showing a large, infiltrative, white-tan lesion. |
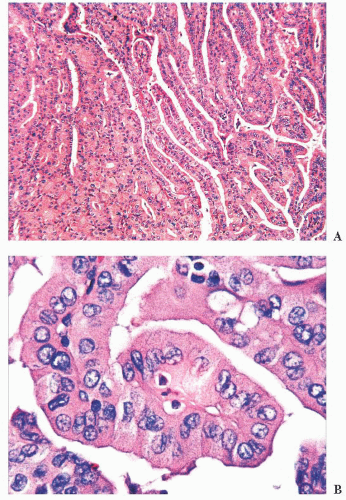 FIGURE 13.17 Tall cell variant of papillary carcinoma. Lowpower view showing a papillary architecture (A) and high-power view showing large elongated tumor cells (cell height three times the width), abundant eosinophilic cytoplasm, and nuclear changes of papillary carcinoma (B). |
from other papillary carcinomas because this lesion is associated with an extremely poor outcome, with most deaths occurring within 5 years of diagnosis (228,230). Grossly, the tumors often measure more than 6 cm. The tumor is characterized microscopically by papillary growth. Tall columnar cells line the papillae. The nuclear features are usually not those of typical papillary carcinomas. The nuclei are hyperchromatic with a punctate chromatin; nuclear stratification is a prominent feature. The cells usually have scant cytoplasm which can be clear. Mitoses are frequently seen. Psammoma bodies are rare. Extrathyroidal extension is common as are distant metastases (230). Encapsulated variants, which may have a better prognosis, have been described (231). Because of its aggressive behavior, the actual classification of this lesion as a variant of papillary carcinoma has been controversial, although most authors include this lesion in the papillary carcinoma classification (230). CDX2, a nuclear transcription factor important in intestinal development, has been shown to be selectively expressed in 50% of cases of CCV (232,233). An encapsulated variant of columnar cell carcinoma has been described, which behaves similarly to conventional PTC (229,234). BRAF mutations can be seen in approximately 33% of CCV-PTC similar to conventional PTC (235).
frequency of LOHs of 3p24, 9p21, 17q21, 21q22, and 22q13 was noted. It has been suggested that these mutations may play an important role in the different morphologic appearance and worse prognosis of DSV.
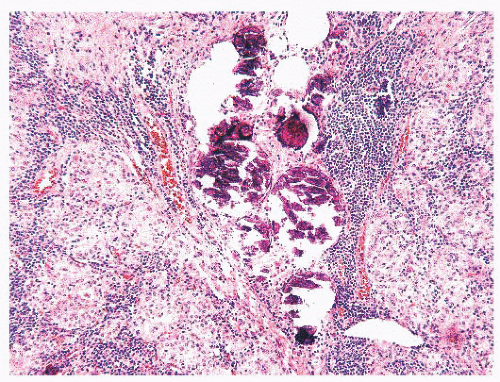 FIGURE 13.18 Diffuse sclerosis variant showing solid tumor nests and numerous psammoma bodies. Notice the lymphocytic infiltrates around the tumor foci. |
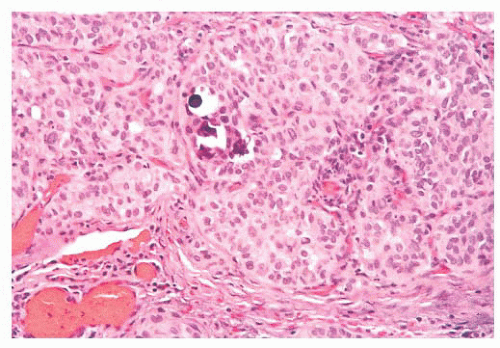 FIGURE 13.19 Solid variant of papillary carcinoma with focal calcifications and psammoma bodies. |
This can be helpful to differentiate these from papillary carcinoma in diagnostically challenging cases (262).
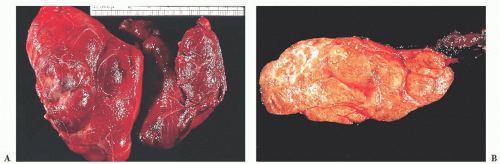 FIGURE 13.20 (A) Multinodular goiter showing asymmetric enlargement of thyroid lobes. (B) Cut section of multinodular goiter showing multiple tan nodules with focal fibrosis. |
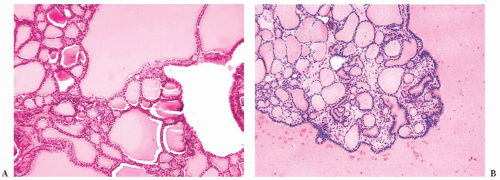 FIGURE 13.21 (A) Papillary hyperplastic nodule showing papillae lined by small round follicular cells and subfollicle formation. (B) Low-power microscopic view of multinodular goiter showing multiple nodules of thyroid follicles of varying size. |
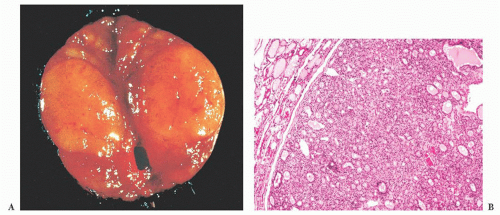 FIGURE 13.22 (A) Gross follicular adenoma showing a tan-brown circumscribed nodule. (B) A low-power view of follicular adenoma showing thinly encapsulated, microfollicular pattern tumor with no capsular or vascular invasion. |
rearrangements in both tumors (288,289). However, to date, no BRAF mutations have been seen in these tumors, and a benign behavior has thus far been described in all cases of hyalinizing trabecular adenoma (290). Therefore, until metastatic behavior is described in a case of hyalinizing trabecular adenoma, these tumors can be designated as hyalinizing trabecular neoplasm.
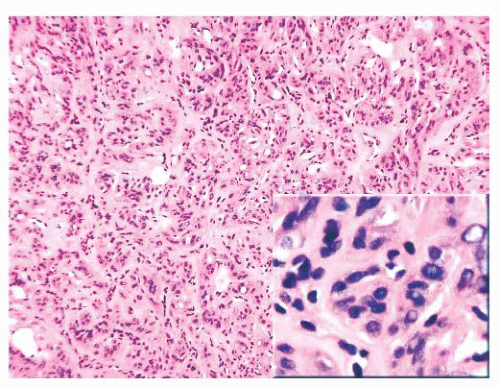 FIGURE 13.23 Histologic examination of a hyalinizing trabecular adenoma shows multiple nests of thyroid follicular epithelium surrounded by a vascular network. The appearances are similar to those seen in a paraganglioma; however, the epithelial cells are derived from follicular epithelium. Note the nuclei appear similar to those of papillary carcinoma (inset). |
appears warranted in those individuals who present with metastatic disease and in whom the primary is still untreated (302). Certainly, lymph node dissection is not warranted because these tumors do not spread to nodes (302).
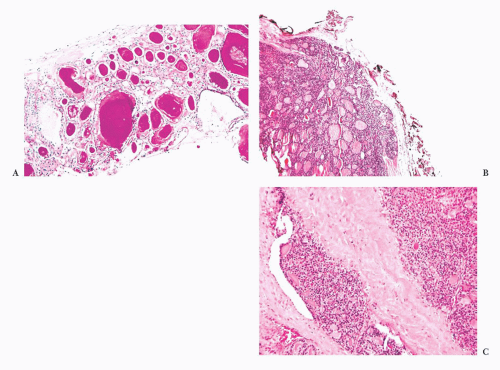 FIGURE 13.24 (A) A bone marrow core biopsy showing replacement of the marrow space by thyroid follicles. This patient had a follicular carcinoma of the thyroid removed 20 years prior to the marrow biopsy. (B) A low-power view of follicular carcinoma showing capsular and vascular invasion. (C) Follicular carcinoma showing vascular invasion. Note the tumor thrombus in the capsular vessels. |
in 90% to 100% of follicular carcinomas and not adenomas. However, others have reported HBME-1 expression in adenomatoid nodules and follicular adenomas (151,286,319).
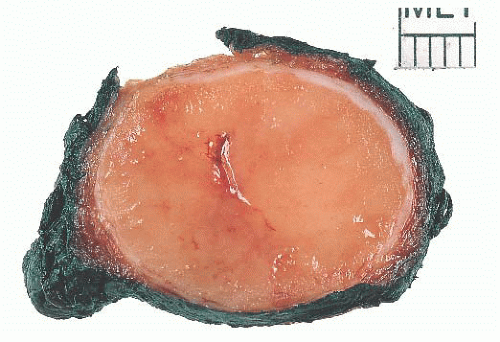 FIGURE 13.25 Gross Hürthle cell adenoma showing a circumscribed orange-brown lesion. Diagnosis relies on histologic examination with thorough examination of the capsule. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


