Basic Considerations

Therapeutic Relevance
CLINICAL SETTING
Those patients who suffer from chronic ailments such as diabetes and epilepsy may have to take drugs every day for the rest of their lives. At the other extreme are those who take a single dose of a drug to relieve an occasional headache. The duration of drug therapy is usually between these extremes. The manner in which a drug is taken is called a dosage regimen. Both the duration of drug therapy and the dosage regimen depend on the therapeutic objective, which may be the cure, the mitigation, or the prevention of disease or conditions suffered by patients. Because all drugs exhibit some undesirable effects, such as drowsiness, dryness of the mouth, gastrointestinal irritation, nausea, and hypotension, successful drug therapy is achieved by optimally balancing the desirable and the undesirable effects. To achieve optimal therapy, the appropriate “drug of choice” must first be selected. This decision implies an accurate diagnosis of the disease or condition, a knowledge of the clinical state of the patient, and a sound understanding of the pharmacotherapeutic management of the disease. Then the questions of, how much? how often? and how long? must be answered.
The question, how much? recognizes that the magnitudes of the therapeutic and adverse responses are functions of the size of dose given. To paraphrase the 16th-century physician Paracelsus, “all drugs are poisons, it is just a matter of dose.” For example, 25 mg of aspirin does little to alleviate a headache; a dose closer to 300 to 600 mg is needed, with little ill effect. However, 10 g taken all at once can be fatal, especially in young children. The question, how often? recognizes the importance of time, in that the magnitude of the effect eventually declines with time following a single dose of drug. The question, how long? recognizes that some conditions are of limited duration, while a cost (in terms of side effects, toxicity, economics) is incurred with continuous drug administration. In practice, these questions cannot be completely divorced from one another. For example, the convenience of giving a larger dose less frequently may be more than offset by an increased incidence of toxicity.
What determines the therapeutic dose of a drug and its manner and frequency of administration, as well as the events experienced over time by patients on taking the recommended dosage regimens, constitutes the body of this book. It aims to demonstrate that there are concepts common to all drugs and that, equipped with these concepts, not only can many of the otherwise confusing events following drug administration be rationalized, but also the key questions surrounding the very basis of dosage regimens can be understood. The intended result is a better and safer use of drugs for the treatment or amelioration of diseases or conditions suffered by patients. Bear in mind, for example, that still today, some 5% of patients admitted into hospital are there because of the inappropriate use of drugs, much of which is avoidable.
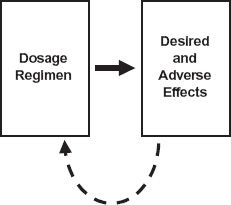
FIGURE 1-1. An empirical approach to the design of a dosage regimen. The effects, both desired and adverse, are monitored after the administration of a dosage regimen of a drug and used to further refine and optimize the regimen through feedback (dashed line).
It is possible, and indeed it was a common past practice, to establish the dosage regimen of a drug through trial and error by adjusting such factors as the dose and interval between doses and observing the effects produced as depicted in Fig. 1-1. A reasonable regimen might eventually be established but not without some patients experiencing excessive adverse effects and others ineffective therapy. Certainly, this was the procedure followed to establish that digoxin needed to be given at doses between 0.125 mg and 0.25 mg only once a day for the treatment of congestive cardiac failure, whereas morphine sulfate needed to be administered at doses between 10 mg and 50 mg up to 6 times a day to adequately relieve the chronic severe pain experienced by patients suffering from terminal cancer. However, this empirical approach not only fails to explain the reason for this difference in the regimens of digoxin and morphine but also contributes little, if anything, toward establishing the principles underlying effective dosage regimens of other drugs. That is, our basic understanding of drugs has not been increased.
INPUT–RESPONSE PHASES
Progress has only been forthcoming by realizing that concentrations at active sites, rather than dose administered, drive responses, and that to achieve and maintain a response, it is necessary to ensure the generation of the appropriate exposure–time profile of drug within the body, which in turn requires an understanding of the factors controlling this exposure profile. These ideas are summarized in Fig. 1-2, where now the input–response relationship (which often runs under the restrictive title “dose–response,” as not only dose is altered) is divided into two parts, a pharmacokinetic phase and a pharmacodynamic phase both with roots derived from the Greek word pharmacon, meaning a drug, or interestingly, a poison. The pharmacokinetic phase covers the relationship between drug input, which comprises such adjustable factors as dose, dosage form, frequency and route of administration, and the concentration achieved with time. The pharmacodynamic phase covers the relationship between concentration and both the desired and adverse effects produced with time. In simple terms, pharmacokinetics may be viewed as how the body handles the drug, and pharmacodynamics as how the drug affects the body. Sometimes, the metabolites of drugs formed within the body have activity and also need to be taken into account when relating drug administration to response.
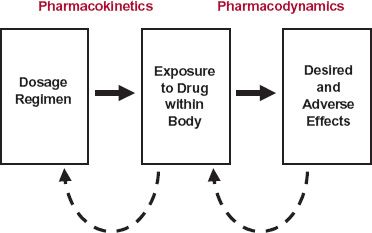
FIGURE 1-2. A rational approach to the design of a dosage regimen. The pharmacokinetics and pharmacodynamics of the drug are first defined. Then, responses to the drug, coupled with pharmacokinetic information, are used as a feedback (dashed lines) to modify the dosage regimen to achieve optimal therapy. For some drugs, active metabolites formed in the body may also need to be taken into account.
Several other basic ideas have helped to place drug administration on a more rational footing. The first, and partially alluded to above, is that the intensity or likelihood of effect increases with increasing exposure to the drug, but only to some limiting, or maximum value, above which the response can go no higher, regardless of how high the exposure. Second, drugs act on different parts of the body, and the maximum effect produced by one drug may be very different from that of another, even though the measured response is the same. For example, both aspirin and morphine relieve pain, but whereas aspirin may relieve mild pain, it cannot relieve the severe pain experienced by patients with severe trauma or cancer even when given in massive doses. Here, morphine, or another opioid analgesic, is the drug of choice. Third, which follows in part from the second idea, is the realization that drugs produce a multiplicity of effects, some desired and others undesired, which when coupled with the first idea, has the following implication. Too low an exposure results in an inadequate desired response, whereas too high an exposure increases the likelihood and intensity of adverse effects. Expressed differently, there exists an optimal range of exposures between these extremes, the therapeutic window, shown schematically in Fig. 1-3. For some drugs, the therapeutic window is narrow, and therefore the margin of safety is small. For others, it is wide. Although the appropriate concentration should be that at the site of action, rarely it is accessible; instead, the concentration is generally measured at an alternative and more accessible site, the plasma.
Armed with these simple ideas, it is now possible to explain the reason for the difference in the dosing frequency between digoxin and morphine. Both drugs have a relatively narrow therapeutic window, but whereas morphine is eliminated very rapidly from the body, so that it must be given frequently to maintain an adequate concentration to ensure relief of pain without excessive adverse effects, such as respiratory depression, digoxin is relatively stable and so with little lost each day, once daily administration suffices. These principles also help to explain an added feature of digoxin. Given daily, digoxin was either ineffective acutely or eventually produced unacceptable toxicity when a dosing rate sufficiently high to be effective acutely was maintained. Because it is eliminated slowly with little lost each day, it accumulates appreciably with repeated daily administration, as depicted schematically in Fig. 1-4. At low daily doses, the initial concentration is too low to be effective but eventually, it rises to within the therapeutic window. Increasing the daily dose brings the concentration within the therapeutic window earlier, but with the concentration still rising, eventually the concentration becomes too high, and unacceptable toxicity ensues. However, what was needed was rapid achievement and subsequent maintenance of adequate digoxin concentrations without undue adverse effects. The answer was to give several small doses within the first day, commonly known collectively as a digitalizing dose, to rapidly achieve therapeutic concentrations followed by small daily doses to maintain the concentration within the therapeutic window. The lesson to be learned from the case of digoxin and indeed most drugs is that only through an understanding of the temporal events that occur after the drug’s administration can meaningful decisions be made regarding its optimal use.
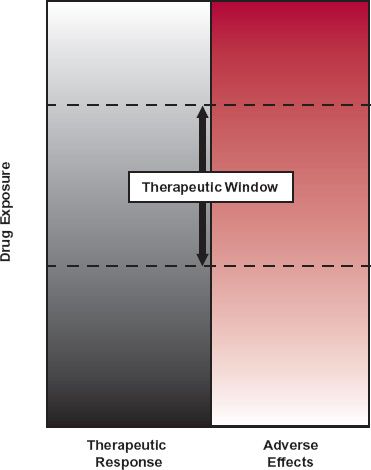
FIGURE 1-3. At higher concentrations or higher rates of administration on chronic dosing, the probability of achieving a therapeutic response increases (from gray to white), but so does the probability of adverse effects (toward increasing redness). A window of opportunity, called the “therapeutic window,” exists, however, within which the therapeutic response can be attained without an undue incidence of adverse effects.
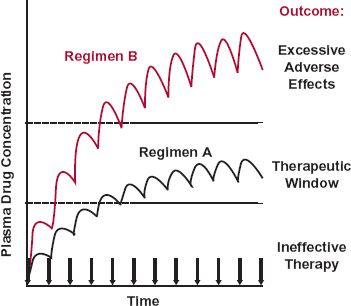
FIGURE 1-4. When a drug is given in a fixed dose and at fixed time intervals (denoted by the arrows), it accumulates within the body until a plateau is reached. With regimen A, therapeutic success is achieved although not initially. With regimen B, the therapeutic objective is achieved more quickly, but the drug concentration is ultimately too high, resulting in excessive adverse effects.
The issue of time delays between drug administration and response is not confined to pharmacokinetics but extends to pharmacodynamics too. Part of such delays is a result of the time required for drug to distribute to the target site, which is often in a cell within a tissue or organ, such as the brain. This is certainly the case with digoxin, where the peak effect on the heart occurs several hours after observing the peak exposure in plasma. Part is also a result of delays within the affected system within the body, as readily seen with the oral anticoagulant warfarin, used as a prophylaxis in the prevention of deep vein thrombosis and other thromboembolic complications. Even though the drug is rapidly absorbed, yielding high, early concentrations throughout the body, as seen in Fig. 1-5, the peak effect, in terms of prolongation of the clotting time, occurs approximately 2 days after a dose of warfarin. Clearly, it is important to take this lag in response into account when deciding how much to adjust dose to achieve and maintain a given therapeutic response. Failing to do so and attempting to adjust dosage based, for example, on the response seen after 1 day, before the full effect develops, increases the danger of overdosing the patient, with serious potential consequences, such as internal hemorrhage, with this low margin-of-safety drug.
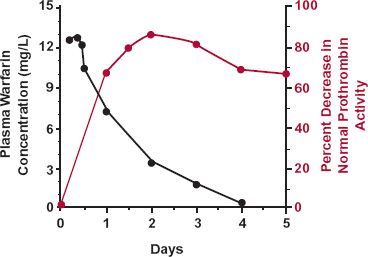
FIGURE 1-5. The sluggish response in the plasma prothrombin complex activity (colored line), which determines the degree of coagulability of blood, is clearly evident following administration of the oral anticoagulant warfarin. Although the absorption of this drug into the body is rapid, with a peak concentration seen within the first few hours, for the first 2 days after giving a single oral 1.5 mg/kg dose of sodium warfarin, response (defined as the percent decrease in the normal complex activity) steadily increases reaching a peak after 2 days. Thereafter, the response declines slowly as absorbed drug is eliminated from the body. The data points are the averages of five male volunteers. (From: Nagashima R, O’Reilly RA, Levy G. Kinetics of pharmacologic effects in man: The anticoagulant action of warfarin. Clin Pharmacol Ther 1969;10:22–35.)
Another example of a time delay in pharmacodynamics concerns an adverse effect, a safety issue, seen in Fig. 1-6
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


