Chapter 9 The role of medicinal chemistry in the drug discovery process
Introduction
Clinically the unmet need for novel and safe drugs is becoming more significant due to increasing longevity in the developed world and the consequential increased prevalence of age-related diseases such as cancer and dementia. Whilst few would dispute that the role of the medicinal chemist is pivotal to the discovery of new medicines to address this growing need, and will continue to be so for the foreseeable future, the environment in which the medicinal chemist operates has changed dramatically over the last 5–10 years. Despite many years of rising investment in R&D, the productivity of major Pharma, in terms of New Chemical Entities (NCEs) delivered to the patient, has not improved over the past 10 years. In 2009 only nineteen NCEs were approved by the FDA, two less than in 2008 (Hughes, 2010) (Figure 9.1; see also Chapter 22). The approval of six biological licence applications in 2009, compared with three in 2008, is perhaps the first indication of the greater emphasis which Pharma is placing on the discovery of biological therapeutic agents. However, the challenges and current limitations of such treatments are such that small molecule discovery is likely to remain the mainstay of therapeutic innovation for at least the next 20 years (but see also Chapter 22).
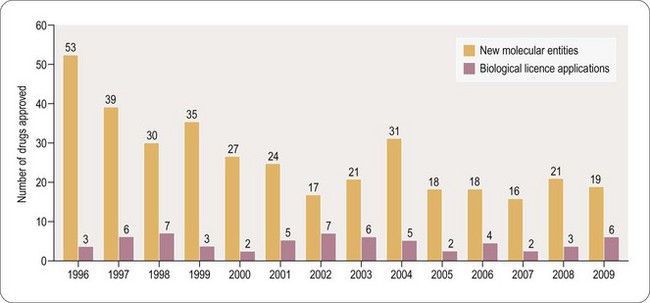
Fig. 9.1 Recent trends in small molecule approvals and biological licience applications –
reproduced with the kind permission of Nature publishing.
Whilst a detailed discussion of the reasons behind Pharma’s poor level of productivity is beyond the scope of this chapter (see Chapter 22), it is clear that the high level of attrition in the clinic, due to lack of clinical efficacy and insufficient safety margins, is a major component and it is appropriate to consider what role the medicinal chemist might play in addressing this challenge in the future. The high level of attrition in the clinic due to lack of sufficient efficacy reflects an inadequate understanding of disease pathophysiology in man and the poor predictability of many of the preclinical animal models of human disease. Thus there is a pressing need to improve the quality of validation of novel targets, placing less emphasis on preclinical in vivo models and single gene ‘knock-outs’, and a greater emphasis on cellular pathways, genetically associated targets and innovative clinical trial design. This approach will require the availability of selective chemical tools with which to validate targets in human tissue and thus the role of the medicinal chemist in the future may incorporate a greater component of chemical biology. The increasing trend towards academic target and drug discovery is also likely to place greater emphasis on chemical biology skills.
Target selection and validation

A key challenge in increasing the industry’s productivity will be the selection of targets which have compelling validation in terms of both efficacy and safety in the human context and, which, consequently, have a greater probability of achieving a successful clinical proof of concept. The role of the medicinal chemist has previously focused primarily on ensuring the quality of preclinical candidate molecules in terms of potency, selectivity and physicochemical properties such as those highlighted initially by Lipinski et al. (Lipinski CA, 1997) and, more recently, by others (Leeson PD and Springthorpe B, 2007). As a result, the level of attrition due to poor ADME properties fell dramatically between 1991 and 2000 (Kola I and Landis J, 2004), but the level of clinical success with new mechanisms remains poor. However, chemists are now increasingly becoming engaged, along with their biology and clinical counterparts, in the process of selecting and prioritizing future protein targets. In particular the development of an in-depth understanding of the structural biology of the target and the biophysics of its interaction with low-molecular-weight ligands is a critical component at the outset of a project and very much in the chemist’s domain. The necessity for a rigorous analysis of potential targets cannot be over-emphasized since this occurs at the start of the 12–15-year period of intensive effort and investment required to achieve the launch of a new medicine into large scale clinical usage. The post genomic era has provided the industry with a plethora of potential drug targets; however, the selection of tractable targets with a high probability of delivering safe and effective treatments represents a huge challenge requiring a multidisciplinary approach.
Whilst a detailed discussion of the complex facets of target validation is beyond the scope of this chapter (see Chapter 6), it is clear that targets with strong genetic associations, such as the voltage-gated sodium channel NaV1.7 (Dib-Hajj SD et al., 2007), with potential utility in the treatment of pain, and the chemokine CCR5 receptor (Westby M and van der Ryst E, 2005), which has led to the discovery of treatments for HIV infection, exemplify an aspirational level of target validation.
Frye has recently described (Frye SV, 2010) essential features of a quality chemical probe which are summarised below:
Molecular profiling. Sufficient in vitro potency and selectivity data to confidently associate its in vitro profile to its cellular or in vivo profile.
Mechanism of action. Activity in a cell-based or cell-free assay influences a physiologic function of the target in a dose-dependent manner.
Identity of the active species. Has sufficient chemical and physical property data to interpret results as due to its intact structure or a well-characterized derivative.
Proven utility as a probe. Cellular activity data available to confidently address at least one hypothesis about the role of the molecular target in a cell’s response to its environment.
Availability. Is readily available to the academic community with no restrictions on use.
It is well known that target classes are associated with different levels of tractability with respect to the probability of being able to identify quality lead molecules. For example the G-protein coupled receptor (GPCR) super-family is generally considered to be one of the most tractable target classes with approximately 40% of prescription drugs in current practice producing efficacy via modulation of a GPCR. In contrast, voltage-gated ion channels are generally regarded as one of the more challenging variety of protein targets to modulate, particularly with respect to subunit specificity. Progress in this area was for many years limited by lack of suitable screening platforms to facilitate drug discovery; however, recent technological developments offer the potential to more fully explore this target class (Kaczorowski GJ et al., 2008).
Various criteria for the selection and prioritization of targets are utilized within major Pharma in the process of building sustainable portfolios of viable potential targets for the discovery scientists to address. A recent illustration (Wehling M, 2009) has highlighted one such approach which objectively applies weighted scores to available target information.
With the emergence of exciting new insights into disease pathologies, the importance of rigorous target selection will become even more important in the future. The availability of the human genome sequence coupled with impressive advances in biology is uncovering challenging targets for the medicinal chemist to address. As an example, epigenetic phenomena are increasingly being recognized as a potentially important area in the development of chronic diseases (Gluckman PD et al., 2008) and the discovery and characterization of specific histone-modifying enzyme subfamilies (Cole PA, 2008) offers the medicinal chemist the opportunity to design specific enzyme modulators with which to regulate gene expression. The availability of selective chemical probes for specific domains within these enzyme classes will be a critical factor in the identification and selection of the most relevant targets. The promise of this area is illustrated by the successful discovery of the histone deacetylase (HDAC) inhibitor Vorinostat (McGuire C and Lee J, 2010; Figure 9.2).
Historically natural products have been considered as a source of both new drugs and potential targets. More recently this approach has suffered demise due to its disappointing productivity. However, there have been a number of recent success stories which are illustrated by the following examples. Ziconotide (Prialt®); (Schmidtko A et al., 2010) is a potent and selective N-type calcium channel blocker approved by the FDA in December 2004 for the treatment of severe chronic pain. Ziconotide has subsequently demonstrated efficacy in patients with refractory pain, i.e. pain which has proven difficult to control using conventional analgesics. Ziconotide is a synthetic peptide derived from a toxin extracted from the marine snail Conus magus. The success of ziconotide has highlighted the potential of natural toxins in drug discovery and has stimulated renewed interest from a number of groups in the area (Halai R and Craik DJ, 2009). Peptides offer high levels of both potency and selectivity which make them valuable tools for target validation and the toxins of a number of venomous species have proved to be a rich source of such molecules. The identification of the endogenous peptide hormone ghrelin (Helstroem PM, 2009), and subsequent understanding of its function, has highlighted the potential clinical value of ghrelin receptor agonists in a number of conditions such as gastroparesis (Ejskjaer N et al., 2009) and postoperative ileus (Popescu I, Fleschner PR et al., 2010) in which the ghrelin receptor agonist TZP-101 (Figure 9.3) has recently demonstrated clinical efficacy. Several of the major disease challenges facing society, such as Alzheimer’s disease, cancer and heart disease, etc., have been shown to be associated, at least in part, with aberrant protein folding and/or protein–protein interactions and the identification of both small molecule and peptidic therapeutics to address these pathologies will require significant innovation.
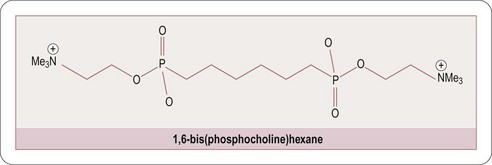
Encouragingly great strides have recently been made in our understanding of the factors which cause proteins to misfold, primarily through the use of biophysical and computational techniques that enable systematic and quantitative analysis of the effects of a range of different perturbations in proteins (Luheshi LM et al., 2008). Recent advances in the design of small molecule inhibitors which inhibit protein–protein interactions has been amply demonstrated by the discovery of the serum amyloid P cross-linking agent CPHPC (Pepys MB et al., 2002; Figure 9.4) and the C-reactive protein inhibitor 1,6-bis(phosphocholine)-hexane (Pepys MB et al., 2006; Figure 9.5).
Lead identification/generation

The identification of high-quality lead molecules is a critical phase in the discovery of drug candidates since decisions made at this point in the process are likely to have a significant impact on the outcome of the project as it sets the framework for future lead optimization and development (Bleicher KH et al., 2003).
Several strategies have been adopted to identify these early hits as listed below:
The widespread use of high-throughput screening has generally been reported to have enabled the identification of leads for approximately 50% of the projects addressed by this approach (Fox S et al., 2006), although a more recent review suggests that, over the past 3 years, the success rate may have increased to approximately 60% (Macarron R et al., 2011). However, it is noteworthy that, despite the massive growth in the numbers of compounds screened over the past 20 years, no corresponding increase in the number of successful registered NCEs has resulted. It has been argued that, in view of increasing development times, it is still too early to evaluate the true success of high-throughput screening (Macarron R et al., 2011). Nevertheless, there has been greater focus on improving the ‘drug and lead-like’ properties and the structure/property diversity of screening collections. ‘Drug-likeness’ can be broadly defined as the overall profile of biophysicochemical properties of a molecule which facilitate its access to and effective mode of interaction at the site of action at a biologically relevant and safe concentration for sufficient duration to elicit the desired therapeutic effect. Leeson and Springthorpe discuss the importance of considering the properties conferring ‘drug-likeness’, particularly lipophilicity, in a recent survey of selected development candidates in leading drug companies (Leeson PD and Springthorpe B, 2007). An analysis of the structural relationship of launched drugs to their corresponding lead molecules revealed that in general the structure of the marketed molecule was very closely related to the lead series (Proudfoot JR, 2002). Thus one could infer that the apparent disappointing success rate of high-throughput screening has been related to the lack of ‘drug likeness’ in the compound collections and significant effort is now being devoted to address this.
Major companies have, therefore, filtered their screening collections to exclude compounds which do not possess physicochemical properties that are generally accepted to confer ‘drug-like’ attributes (Bleicher KH et al., 2003). However, it should be noted that, although the discovery of drug-like preclinical development candidates requires the initial identification of quality leads, the properties which confer ‘drug-likeness’ and those associated with ‘lead-likeness’ are not the same (Rishton GM, 2003). It is, therefore, important to understand the components conferring ‘lead-likeness’ when assessing screening collections. A publication from the Astra-Zeneca group addressed the design of lead-like combinatorial libraries and suggested that leads suitable for further optimization are most likely to be relatively polar, low molecular weight (MW = 200–350) and of relatively low lipophilicity (clogP ca.1.0–3.0) (Oprea TI et al., 2001). The authors point out that when beginning from such a tractable low-molecular-weight lead molecule, subsequent optimization to increase potency and selectivity is likely to increase molecular weight and lipophilicity to values in the order of the accepted ‘drug-likeness’ parameters.
High-throughput screening (Chapter 8) continues to be a valuable strategy where there is little information around the structure of the biological target or its ligands; however, where such knowledge exists, it has generally been more successful to utilize this information to identify potential molecular starting points. For example, structural information on ligands which are known to interact with a target can be used to construct a pharmacophore, which may be used in an initial virtual screening programme of proprietary or commercial compound collections to enrich the sample set of compounds ultimately selected for actual screening. A critical review assessing the past effectiveness of virtual screening and proposing considerations for future improvements has recently appeared (Schneider G, 2010). Computational tools are widely used to generate in silico pharmacophores, using either molecular or electronic overlays. The introduction of electronic field overlay technology has allowed chemists to interrogate electronic properties in a relatively accessible manner. Similarly the latter technology may be usefully applied to ‘scaffold hopping’ where a structurally distinct compound class has been derived from an active series via application of a field pharmacophore (Cheeseright TJ et al., 2008). An extreme example of utilizing structural information is where an existing known drug molecule is used as the starting point to discover an improved agent (Naik P et al., 2010).
Although the majority of high-throughput screens are aimed at known biological targets with supporting target validation in the disease of interest, there has been significant activity in establishing screens based on the cell phenotype, i.e. where biochemical pathways are targeted as opposed to specific proteins within the pathway. The major challenge associated with this approach is the subsequent identification of the specific molecular target or targets with which the agents interact to produce the desired cellular response (Hart CP, 2005). Whilst in some cases further optimization of leads has been carried using the phenotypic assay itself, because of the numerous variables involved, e.g. multiple targets and cellular distribution, etc., precise SAR can be difficult to obtain from the phenotypic assay. Thus a detailed analysis and elucidation of the mechanism of action is often required to enable rapid progress to be achieved using a protein specific assay.
Over the past few years fragment-based drug discovery has become an established technique for lead generation, often in conjunction with other strategic approaches (Whittaker M et al., 2010). Fragment-based screening consists of screening a chemical library of low-molecular-weight compounds, usually at high concentration, using sensitive assay methods. In addition to biochemical assays such campaigns often incorporate biophysical methodology such as NMR, surface plasmon resonance (SPR), isothermal titration calorimetry (ITC) and X-ray crystallography (Carr R and Jhoti H, 2002; Rees DC et al., 2004; Orita M et al., 2009). The considerable progress achieved with in silico design of fragment libraries using a variety of techniques has recently been reviewed (Konteatis ZD, 2010). Essentially the fragment-based approach aims to identify ligands for the target protein which have relatively low affinity but high ligand/ligand-lipophilicity efficiency. Ligand efficiency (LE) is a measure of the binding energy of a molecule normalized by its size. This is often calculated by dividing the pKi or IC50 by the molecular weight or number of heavy atoms. Similarly ligand-lipophilicity efficiency (LLE) can be calculated by determining the difference between the pKi or pIC50 value and the clogP, which provides a measure of the binding energy of a molecule normalized by its lipophilicity (Smith GF, 2009). Thus compounds with high LE and LLE interact highly efficiently with the biological target. Subsequent optimization is then focused on increasing potency whilst maintaining high LE/LLE, thus avoiding the pitfall of increasing potency by increasing size and lipophilicity which often leads to promiscuous ‘off-target’ interactions and subsequent toxicity. Although the concepts of LE and LLE have been particularly applied in fragment-based design they are now widely applied across all lead generation strategies. There are now numerous examples of a fragment-base approach being employed in drug discovery projects and several have been reviewed recently (Congreve M, Chessari G et al., 2008).
In stark contrast to the fragment-based approach to lead generation, natural products have been, and arguably continue to be, a fruitful source of lead molecules and drugs over the past 25–30 years (Newman DJ and Cragg JM, 2007). However, despite the demonstrable historical successes of natural product research, major Pharma has largely reduced its investment in natural product screening in favour of the small molecule library approaches described above. The anticipation that combinatorial chemistry/automated synthesis would provide an excess of small molecule leads for most biological targets was partly responsible for the demise of this strategy. Furthermore the technical challenges associated with the deconvolution of multicomponent natural product mixtures and subsequent chemical simplification of large and complex structures has also deterred continued investment in this field. Nevertheless a number of smaller companies have recognized this as an opportunity and are pursuing this strategy for lead identification and it has recently been argued that the technical challenges associated with this approach have been lessened (Harvey AI, 2008; Harvey AI, 2010).
Despite the relative demise of natural product screening over the past few years, natural products have inspired the development of new strategies in the identification of privileged structures from which small molecule libraries have been constructed. Herbert Waldmann has received notable acclaim for his biology-oriented synthesis (BIOS) strategy which encompasses the development of small molecular entities derived from natural product and drug families, the so-called ‘structural classification of natural products’ (SCONP) (Keiser M et al., 2008) and an associated ‘scaffold hunter’ approach (Wetzel S et al., 2009).
X-ray crystallography has been effectively applied over many years to optimize small molecule ligands primarily for soluble enzyme targets. With the introduction of fragment-based screening high-throughput X-ray crystallography has over the past decade also assumed an important role in the identification of lead molecules (Carr R and Jhoti H, 2002). Similarly, NMR is proving to be an effective, accessible, complementary technique in identifying ligand-protein interactions particularly for membrane bound proteins such as GPCRs, for which detailed X-ray structures are relatively rare (Bartoschek S et al., 2010).
Recent developments in the acquisition of bioengineered protein and subsequent X-ray crystal structures of stabilized GPCRs (Tate GC and Schertler GFX, 2009) may provide exciting future opportunities for medicinal chemists to actively use ligand-bound co-crystal structures of GPCRs in an analogous fashion to the enzyme co-crystallization structures which have greatly facilitated the discovery of selective enzyme inhibitors.
Industry pressure to improve productivity and efficiency in the identification of new chemical entities has added stimulus to the exploration of new technologies in the identification of lead molecules. In particular miniaturization and automation of chemical synthesis and biological screening assays are currently under intense focus (Lombardi D and Dittrich PS, 2010).
The identification of screening platforms which obviate the requirement for artificial labels or reporter systems is also receiving considerable attention (Shiau AK et al., 2008). Biophysical methods such as surface-plasmon resonance and impedance-based technologies are developing rapidly and will become more widely used, particularly since these approaches are less likely to identify false positives which result from labelling artefacts and aggregation phenomena (Giannetti AM et al., 2008). The successful development of these new technologies has the potential to transform the lead identification process and address, in part, the challenge of improving productivity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


