Chapter 7 The role of information, bioinformatics and genomics
The pharmaceutical industry as an information industry
As outlined in earlier chapters, making drugs that can affect the symptoms or causes of diseases in safe and beneficial ways has been a substantial challenge for the pharmaceutical industry (Munos, 2009). However, there are countless examples where safe and effective biologically active molecules have been generated. The bad news is that the vast majority of these are works of nature, and to produce the examples we see today across all biological organisms, nature has run a ‘trial-and-error’ genetic algorithm for some 4 billion years. By contrast, during the last 100 years or so, the pharmaceutical industry has been heroically developing handfuls of successful new drugs in 10–15 year timeframes. Even then, the overwhelming majority of drug discovery and development projects fail and, recently, the productivity of the pharmaceutical industry has been conjectured to be too low to sustain its current business model (Cockburn, 2007; Garnier, 2008).
A great deal of analysis and thought is now focused on ways to improve the productivity of the drug discovery and development process (see, for example, Paul et al., 2010). While streamlining the process and rebalancing the effort and expenditures across the various phases of drug discovery and development will likely increase productivity to some extent, the fact remains that what we need to know to optimize productivity in drug discovery and development far exceeds what we do know right now. To improve productivity substantially, the pharmaceutical industry must increasingly become what in a looser sense it always was, an information industry (Robson and Baek, 2009).
Innovation depends on information from multiple sources
Information from three diverse domains sparks innovation in drug discovery and development (http://dschool.stanford.edu/big_picture/multidisciplinary_approach.php):
• Feasibility (Is the product technically/scientifically possible?)
• Viability (Can such a product be brought cost-effectively to the marketplace?)
The viability and desirability domains include information concerning public health and medical need, physician judgment, patient viewpoints, market research, healthcare strategies, healthcare economics and intellectual property. The feasibility domain includes information about biology, chemistry, physics, statistics and mathematics. Currently, the totality of available information can be obtained from diverse, unconnected (’siloed’) public or private sources, such as the brains of human beings, books, journals, patents, general literature, databases available via the Internet (see, for example, Fig. 7.1) and results (proprietary or otherwise) of studies undertaken as part of drug discovery and development. However, due to its siloed nature, discreet sets of information from only one domain are often applied, suboptimally, in isolation to particular aspects of drug discovery and development. One promising approach for optimizing the utility of available, but diverse, information across all aspects of drug discovery and development is to connect apparently disparate information sets using a common format and a collection of rules (a language) that relate elements of the content to each other. Such connections make it possible for users to access the information in any domain or set then traverse across information in multiple sets or domains in a fashion that sparks innovation. This promising approach is embodied in the Semantic Web, a vision for the connection of not just web pages but of data and information (see http://en.wikipedia.org/wiki/Semantic_Web and http://www.w3.org/2001/sw/), which is already beginning to impact the life sciences, generally, and drug discovery and development, in particular (Neumann and Quan, 2006; Stephens et al., 2006; Ruttenberg et al., 2009).
Bioinformatics
• The European Bioinformatics Institute (EMBL-EBI), which is part of the European Molecular Biology Laboratory (EMBL) (http://www.ebi.ac.uk/2can/home.html)
• The National Center for Biotechnology Information of the National Institutes of Health (http://www.ncbi.nlm.nih.gov/Tools/)
• The Biology Workbench at the University of San Diego (http://workbench.sdsc.edu/).
Bioinformatics is the management and data analytics of bioinformation. In genetics and molecular biology applications, that includes everything from classical statistics and specialized applications of statistics or probability theory, such as the Hardy–Weinberg equilibrium law and linkage analysis, to modelling interactions between the products of gene expression and subsequent biological interpretation (for interpretation tool examples see www.ingenuity.com and www.genego.com). According to taste, one may either say that a new part of data analytics is bioinformatics, or that bioinformatics embraces most if not all of data analytics as applied to genes and proteins and the consequences of them. Here, we embrace much as bioinformatics, but also refer to the more general field of data analytics for techniques on which bioinformatics draws and will continue to borrow.
General principles for data mining
Data mining algorithms can yield information about:
• the presence of subgroups of samples within a sample dataset that are similar on the basis of patterns of characteristics in the variables for each sample (clustering samples into classes)
• the variables that can be used (with weightings reflecting the importance of each variable) to classify a new sample into a specified class (classification)
• mathematical or logical functions that model the data (for example, regression analysis) or
• relationships between variables that can be used to explore the behaviour of the system of variables as a whole and to reveal which variables provide either novel (unique) or redundant information (association or correlation).
Some general principles for data mining in medical applications are exemplified in the mining of 667 000 patient records in Virginia by Mullins et al. (2006). In that study, which did not include any genomic data, three main types of data mining were used (pattern discovery, predictive analysis and association analysis). A brief description of each method follows.
Pattern discovery
Pattern discovery is a data mining technique which seeks to find a pattern, not necessarily an exact match, that occurs in a dataset more than a specified number of times k. It is itself a ‘number of times’, say n(A & B & C &…). Pattern discovery is not pattern recognition, which is the use of these results. In the pure form of the method, there is no normalization to a probability or to an association measure as for the other two data mining types discussed below. Pattern discovery is excellent at picking up complex patterns with multiple factors A, B, C, etc., that tax the next two methods. The A, B, C, etc. may be nucleotides (A, T, G, C) or amino acid residues (in IUPAC one letter code), but not necessarily contiguous, or any other variables. In practice, due to the problems typically addressed and the nature of natural gene sequences, pattern discovery for nucleotides tends to focus on runs of, say, 5–20 symbols. Moreover, the patterns found can be quantified in the same terms as either of the other two methods below. However, if one takes this route alone, important relationships are missed. It is easy to see that this technique cannot pick up a pattern occurring less than k times, and k = 1 makes no sense (everything is a pattern). It must also miss alerting to patterns that statistically ought to occur but do not, i.e. the so-called unicorn events. Pattern discovery is an excellent example of an application that does have an analogue at operating system level, the use of the regular expression for partial pattern matching (http://en.wikipedia.org/wiki/Regular_expression), but it is most efficiently done by an algorithm in a domain-specific application (the domain in this case is fairly broad, however, since it has many other applications, not just in bioinformatics, proteomics, and biology generally). An efficient pattern discovery algorithm developed by IBM is available as Teireisis (http://www.ncbi.nlm.nih.gov/pmc/articles/PMC169027/).
Predictive analysis
Predictive analysis encompasses a variety of techniques from statistics and game theory that analyse current and historical facts to predict future events (http://en.wikipedia.org/wiki/Predictive_analytics). ‘Predictive analysis’ appears to be the term most frequently used for the part of data mining that involves normalizing n(A & B & C) to conditional probabilities, e.g.
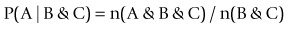 (1)
(1)Association analysis
Association analysis is often formulated in log form as Fano’s mutual information, e.g.
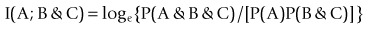 (2)
(2)(Robson, 2003, 2004, 2005, 2008; Robson and Mushlin, 2004; Robson and Vaithiligam, 2010). Clearly it does take account of the occurrence of A. This opens up the full power of information theory, of instinctive interest to the pharmaceutical industry as an information industry, and of course to other mutual information measures such as:
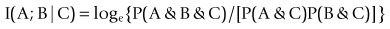 (3)
(3)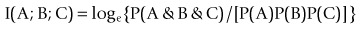 (4)
(4)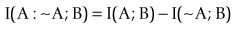 (5)
(5)where ~A is a negative of complementary state or event such that
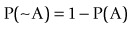 (6)
(6)is familiar as log predictive odds, while
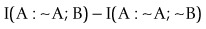 (7)
(7)The above seem to miss out various forms of data mining such as time series analysis and clustering analysis, although ultimately these can be expressed in the above terms. What seems to require an additional comment is correlation. While biostatistics courses often use association and correlation synonymously, data miners do not. Association relates to the extent to the number of times things are observed together more, or less, than on a chance basis in a ‘presence or absence’ fashion (such as the association between a categorical SNP genotype and a categorical phenotype). This is reminiscent of the classical chi square test, but revealing the individual contributions to non-randomness within the data grid (as well as the positive or negative nature of the association). In contrast, correlation relates to trends in values of potentially continuous variables (independence between the variances), classically exemplified by use of Pearson’s correlation. Correlation is important in gene expression analysis, in proteomics and in metabolomics, since a gene transcript (mRNA) or a protein or a small molecule metabolite, in general, has a level of abundance in any sample rather than a quantized presence/absence. Despite the apparent differences, however, the implied comparison of covariance with what is expected on independent, i.e. chance, basis is essentially the same general idea as for association. Hence results can be expressed in mutual information format, based on a kind of fuzzy logic reasoning (Robson and Mushlin, 2004).
Genomics
The genome and its offspring ‘-omes’
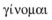 ) means I become, I am born, to come into being, and the Oxford English Dictionary gives its aetiology as being from gene and chromosome. This aetiology may not be entirely correct.
) means I become, I am born, to come into being, and the Oxford English Dictionary gives its aetiology as being from gene and chromosome. This aetiology may not be entirely correct.While the term genome has recently spawned many offspring ‘-omes’ relating to the disciplines that address various matters downstream from inherited information in DNA, e.g. the proteome, these popular -ome words have an even earlier origin in the 20th century (e.g. biome and rhizome). Adding the plural ‘-ics’ suffix seems recent. The use of ‘omics’ as a suffix is more like an analogue of the earlier ‘-netics’ and ‘-onics’ in engineering. The current rising hierarchy of ‘-omes’ is shown in Table 7.1, and these and others are discussed by Robson and Baek (2009). There are constant additions to the ‘-omes’.
Table 7.1 Gene to function is paved with ‘-omes’
| Commonly used terms | ||
| Genome | Full complement of genetic information (i.e. DNA sequence, including coding and non-coding regions) | Static |
| Transcriptome | Population of mRNA molecules in a cell under defined conditions at a given time | Dynamic |
| Proteome | Either: the complement of proteins (including post-translational modifications) encoded by the genome | Static |
| or: the set of proteins and their post-translational modifications expressed in a cell or tissue under defined conditions at a specific time (also sometimes referred to as the translatome) | Dynamic | |
| Terms occasionally encountered (to be interpreted with caution) | ||
| Secretome | Population of secreted proteins produced by a cell | Dynamic |
| Metabolome | Small molecule content of a cell | Dynamic |
| Interactome | Grouping of interactions between proteins in a cell | Dynamic |
| Glycome | Population of carbohydrate molecules in a cell | Dynamic |
| Foldome | Population of gene products classified by tertiary structure | Dynamic |
| Phenome | Population of observable phenotypes describing variations of form and function in a given species | Dynamic |
The new ‘-omes’ are not as simple as a genome. For one thing, there is typically no convenient common file format or coding scheme for them comparable with the linear DNA and protein sequence format provided by nature. Whereas the genome contains a static compilation of the sequence of the four DNA bases, the subject matters of the other ‘-omes’ are dynamic and much more complex, including the interactions with each other and the surrounding ecosystem. It is these shifting, adapting pools of cellular components that determine health and pathology. Each of them as a discipline is currently a ‘mini’ (or, perhaps more correctly, ‘maxi’) genome project, albeit with ground rules that are much less well defined. Some people have worried that ‘-omics’ will prove to be a spiral of knowing less and less about more and more. Systems biology seeks to integrate the ‘-omics’ and simulate the detailed molecular and macroscopic processes under the constraint of as much real-world data as possible (van der Greef and McBurney, 2005; van der Greef et al., 2007).
For the purposes of this chapter, we follow the United States Food and Drug Administration (FDA) and European Medicines Evaluation Agency (EMEA) agreed definition of ‘genomics’ as captured in the definition of a ‘genomic biomarker’, which is defined as: A measurable DNA and/or RNA characteristic that is an indicator of normal biologic processes, pathogenic processes, and/or response to therapeutic or other interventions (see http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073162.pdf). A genomic biomarker can consist of one or more deoxyribonucleic acid (DNA) and/or ribonucleic acid (RNA) characteristics.
DNA characteristics include, but are not limited to:
• Single nucleotide polymorphisms (SNPs);
• Variability of short sequence repeats
• Deletions or insertions of (a) single nucleotide(s)
• Cytogenetic rearrangements, e.g., translocations, duplications, deletions, or inversions.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


