, Mohammed Al-Rubaie2, Stuart Walker3 and Sam Salek4
(1)
Regulatory Affairs Consultant Executive Directive, Kuwait Advancement for Conference and Exhibition Management, Kuwait, Kuwait
(2)
Ministry of Health, Directorate General of Pharmaceutical Affairs and Drug Control, Muscat, Oman
(3)
Founder of Centre for Innovation in Regulatory Science, London, UK
(4)
Department of Pharmacy School Life & Medical Sciences, University of Hertfordshire, Hatfield, UK
Introduction
Modern-day licensing began in the 1940s with the formation and constitution of the World Health Organization (WHO) and its recommendation that global standards be established in relation to the safety, quality and efficacy of biological, pharmaceutical and similar products and extending this to their labelling and advertising (Crout 1998). However, there is little conformity between countries worldwide as to how the review is conducted including what stages comprise the process, who carries out each stage, what criteria are employed, how long it takes or, indeed, whether there is a review process at all. About 30 % of WHO member states either have only a very rudimentary drug regulatory authority or none at all, whilst only 20 % are thought to have a well-developed drug registration system (Ratanawijitrasin and Wondemagegnehu 2002).
This chapter focuses on the Gulf Cooperation Council (GCC) states: Bahrain, Kuwait, Oman, Qatar, Saudi Arabia and UAE. Yemen was not included in the evaluation due to its differences from the six states particularly in relation to its economic status and the development of its regulatory system.
The six GCC regulatory authorities have the same goals in protecting local consumers from the harmful effects of medicines by ensuring the availability of medicinal products of desirable quality, safety and efficacy in each country. However, the practices and strategies involved in carrying out the regulatory review processes vary across the region. From the pharmaceutical industry’s perspective, the regulatory review of new medicines is the culmination of a research and development process that has taken between 12 and 14 years (McAuslane et al. 2004), and estimates about the cost of developing a new drug vary widely, from a low of $800 million to $2.6 billion per drug (Tufts Center for the Study of Drug Development 2014). Therefore, this study uses a structured approach to collect comprehensive data on the regulatory review process across the GCC region. The assessment is based on the argument that, despite the noticeable differences between different regulatory processes, the processes are made up of a set of basic stages sufficiently similar to allow meaningful comparisons (Hirako et al. 2007).
All the GCC authorities have a similar structure when reviewing pharmaceutical product dossiers, but the position of each milestone in the review process differs from one state to another (Hashan 2005). It is recognised that individual authorities have various experiences and knowledge that could be of value to each other through the comparison of various systems and sharing of best practices to the advantage of all. With this in mind, this study was conducted to compare the review practices in the seven GCC states.
This chapter evaluates the key stages in each review process to determine the commonly shared milestones of the regulatory review process across the member states. The key milestones in the approval process, which are recognised and shared in most of the GCC states, are defined in Table 2.1.
Table 2.1
Definitions of key milestones identified in the Gulf Cooperation Council (GCC) regulatory review processes
Review phases | Key milestone | Suggested definition |
|---|---|---|
Submission phase | Receipt stage | The authority may request a pre-submission document for the application to be accepted, for example, notification to submit from the sponsor |
Queuing for review | This is the stage where the received applications are pending for action to begin | |
Validation stage | This may include administrative procedures such as checks on completeness of the dossier to include all the documents required, checks on legal requirements, status of the company, local agent, manufacturer, etc. | |
Evaluation phase | Scientific assessment | The assigned member of the scientific committee or a pharmacist from the department carries out the scientific assessment and generates a report. Sometimes the registration committee assesses the pharmacist’s report and makes the final registration decision. In some systems the clock stops when questions are asked and sponsor time can be measured and deducted from the authority review time |
Questions to sponsor | May be batched and sent at one time or asked throughout the review process, in which case the sponsor time is not easily measured | |
Quality control analysis | The national quality control laboratory analyses the pharmaceutical product as a requirement for registration and generates a report | |
Authorisation phase | Pricing process | All GCC authorities carry out the pricing of products before they are allowed to enter the local market, but they differ in their pricing procedure and the final price approval |
Authorisation process | This is the process after the scientific review whilst the formal authorisation is issued. It may be extended by pricing negotiations and finalisation of analytical and/or GMP checks | |
Approval time | This is the time interval from the submission stage to the final issue of the registration certificate |
The evaluation of the GCC review process was based on data, which were collected on applications for New Active Substances (NAS) and Existing Active Substances (EAS) that had not previously been approved by the authority in question. The review stages and milestones that could be compared across regulatory authorities were identified, in spite of any differences between the individual regulatory procedures.
The six authorities share similar goals, objectives and obligations to safeguard public health when assessing the safety, quality and efficacy of medicines before they are authorised for marketing. This is no exception to any other authority in the world, and in order to achieve this target, each country has laws, strategies and regulations for approving and marketing pharmaceutical products. Therefore, for the purpose of clarity, the assessment is divided into two parts:
Part I: Addresses the regulatory review process in individual Gulf states
Part II: Provides a comparative assessment of the review milestones between the GCC states
Part I: Regulatory Review Processes in the Gulf States
Pharmaceutical companies are obliged to demonstrate evidence of their product’s quality, safety and efficacy standards and must submit data to the regulatory authorities reporting reasonable biological and chemical activities in order to be considered for registration for human use. Further evidence of the product’s registration and marketing in other countries is required prior to making the final approval decision.
Review model(s) for the scientific assessment is determined by the extent to which data is assessed in detail by the authority rather than relying on the results of assessments and reviews carried out elsewhere. Many authorities apply a different level of data assessment to different applications, according to the type of product and/or its regulatory status with other authorities. There are three basic types of assessment models, which were identified and described below.
Verification Model (Type I Assessment)
This model is used to reduce duplication of effort by agreeing that the importing country will allow certain products to be marketed locally once they have been authorised by one or more recognised reference agencies, elsewhere. The main responsibility of the authority in the importing country is to ‘verify’ that the product intended for local sale has been duly registered as declared in the application and that the product characteristics (formulation, composition) and the prescribing information (use, dosage, precautions) for local marketing conform to that agreed in the reference authorisation(s).
Abridged Model (Type II Assessment)
This model also conserves resources by not reassessing scientific supporting data that has been reviewed and accepted elsewhere but includes an ‘abridged’ independent review of the product in terms of its use under local conditions. This might include a review of the pharmaceutical quality (CMC) data in relation to climatic conditions and distribution infrastructure and a benefit-risk assessment in relation to use in the local ethnic population, medical practice/culture and patterns of disease and nutrition.
Full Review Model (Type III Assessment)
In this model the authority has suitable resources, including access to appropriate internal and external experts, to carry out a ‘full’ review and evaluation of the supporting scientific data (quality, preclinical, clinical) for a major application. A type III assessment could be carried out on a new application that has not been approved elsewhere, but in practice legal requirements may dictate that the product must be authorised by a reference authority before the local authorisation can be finalised.
The models of the review process carried out in the GCC states significantly vary according to the respondent’s perceptions and views of their own review practices. The level of data assessment in each authority depends on the type of product and/or its regulatory status with other authorities. The three types of assessment models were explored and the extent of the scientific reviews was examined for each GCC authority.
Five GCC authorities stated that they perform an abridged assessment (Bahrain, Kuwait, Oman, Qatar and UAE). This is a critical practice to ensure the appropriateness of the product under local conditions. Bahrain carries out a verification review for biological and biotech products because they have to be registered in other reference authorities to be accepted for review in Bahrain and an abridged review for other major applications. The UAE conducts an abridged review for biological and biotech products because they are only registered if they are approved by advanced regulatory authorities and conducts a full review for other major applications. Saudi Arabia is the only country that performs a full review for all types of applications (Table 2.2).
Table 2.2
Models of assessment and the extent of the scientific review in the Gulf Cooperation Council (GCC) authorities
Type of review model | Bahrain | Kuwait | Oman | Qatar | Saudi Arabia | UAE |
|---|---|---|---|---|---|---|
Verification review (type I) | ✓ | × | × | × | × | × |
Abridged review (type II) | ✓ | ✓ | ✓ | ✓ | × | ✓ |
Full review (type III) | × | × | × | × | ✓ | ✓ |
Similarity to locally registered product | ||||||
Fully identical | ✓ | ✓ | × | ✓ | ✓ | ✓ |
Mostly identical | × | × | ✓ | × | × | × |
Closely similar | × | × | × | × | × | × |
Extent of scientific review | ||||||
1. Chemistry and Manufacturing Control (CMC) data | ||||||
Detailed review | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
Reviewed when necessary | × | × | ✓ | × | × | × |
2. Nonclinical data | ||||||
Detailed | × | × | × | × | ✓ | ✓ |
Reviewed when necessary | ✓ | ✓ | ✓ | ✓ | × | ✓ |
3. Clinical data | ||||||
Detailed | × | × | × | × | ✓ | ✓ |
Reviewed when necessary | ✓ | ✓ | ✓ | ✓ | × | ✓ |
Additional information obtained from | ||||||
Other agencies’ internal review reports | ✓ | × | × | ✓ | × | × |
Reports available on the Internet | ✓ | ✓ | ✓ | ✓ | × | ✓ |
General Internet search | ✓ | ✓ | ✓ | ✓ | × | ✓ |
Furthermore, the extent of the scientific assessment was evaluated in the six GCC authorities, and the results revealed that six GCC authorities perform detailed assessment on the pharmaceutical quality (CMC) data. The six authorities have assessors with the required skills and experience to evaluate the CMC data. Oman and Kuwait perform detailed assessment on products which are not registered in countries with reference authorities, whilst they review the pharmaceutical quality data for products which are registered in recognised agencies when there is a query to be examined. Detailed nonclinical and clinical assessments are performed by Saudi Arabia and UAE. In UAE, these detailed assessments are performed on products which are not approved by recognised authorities, whilst those which show evidence of authorisation in reference regulatory authorities are only examined when there is a query.
Bahrain, Kuwait, Oman and Qatar perform the nonclinical and clinical assessment when there is a critical issue to be evaluated. This may be due to the lack of expertise to carry out nonclinical and clinical assessment in these four authorities.
Three common phases are thoroughly examined and described for each GCC regulatory review process, namely, the submission phase, the evaluation phase and the authorisation phase. These data reflect the situation at the time the study was carried out (2010) and subsequent changes in the regulatory environment will need to be monitored.
Regulatory Review Process in Bahrain
Bahrain has a unique medicines policy that clearly states the aims, the current situation and the objectives of the Bahraini medicines control system. It is called the ‘Bahrain Medicines Policy – BMP’. The goal of the policy is to serve as a guide for action and commitment to provide good-quality, safe and effective medicines which are rationally used and provided at reasonable costs for the people in Bahrain and for coping with new developments in the field of pharmaceuticals (Bahrain Ministry of Health 2008). The regulatory review process in Bahrain is illustrated in Fig. 2.1 comprising the critical steps that form substantial parts of the review process.
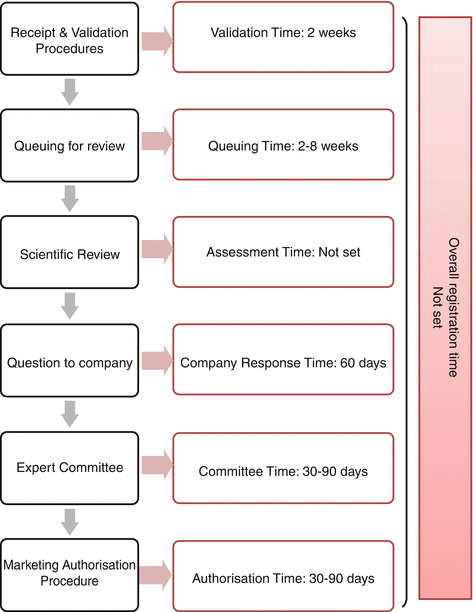
Fig. 2.1
The regulatory review process map for Bahrain
The Submission Phase
Initially, the sponsor submits the product registration dossier with the complete documents for the official acceptance of the dossier to be made. The authority does not specify information about the logistics involved at the receiving stage.
However, the dossier is validated before it is accepted for review, and the aspects such as legal status of applicant/local agent, GMP status of manufacturer, patent/IP status of active ingredients, acceptable format of the application, organised format of the registration dossier including the three sections of scientific data (quality, safety and efficacy) and CPP authenticated by the respective embassy or consulate general are checked accordingly.
The target validation time within the authority is 2 weeks after which it is officially accepted for review once all the missing data has been provided and the date of acceptance is recorded. The Bahraini authority refuses an incomplete application and generates an official letter indicating the missing data and a time period of 2–4 weeks for the application to be completed. The dossier, then, joins the queue for a period of 2–8 weeks before entering the scientific assessment stage. The authority recognises the medical urgency of the priority review process, and therefore emergency, life-saving and important medicines are always taken out of the queue for the accelerated review process.
The Evaluation Phase
When the product enters the scientific assessment stage, the dossier is split into three sections, which are assessed in parallel by the appointed reviewer who completes a product assessment template and collects all the resulting questions as they arise during the review into one batch for the sponsor including the laboratory requirements. After sending the queries, the sponsor is given a time limit of 6 weeks to respond, and all inquiries regarding the product labelling information are negotiated with the sponsor during the evaluation phase. The sponsor holds meetings with the authority’s staff to discuss any questions that arise during the assessment. Finally, the product is sent to the quality control laboratory to carry out sample analysis to determine the eligibility of the product for approval.
The procedures of the scientific committee for the assessment stage are integrated into the authority’s own internal/external scientific review process. The committee’s experts (internal and external) carry out the review process, and the authority is mandated to follow the committee’s recommendations. The time for the committee review is 30–90 days, after which the decision is made to grant the marketing authorisation.
The Authorisation Phase
The pricing process is the final step and price negotiations occur at the end of the scientific assessment. The sponsor is informed of a positive scientific opinion within 90 days of issuing the authorisation. At this point the pricing negotiations and scientific assessment procedures are complete, and the product is ready for approval. The company is now required to pay the fees to receive the registration certificate, and the product is ready to be marketed in Bahrain.
The key milestones in the approval process and target approval time in Bahrain are illustrated in Fig. 2.1. The authority has not set a target time for the scientific assessment stage, and therefore it was not possible to calculate the final product approval time.
Regulatory Review Process in Kuwait
The regulatory review process in Kuwait focuses on the quality review for pharmaceutical products to be authorised for marketing in Kuwait (Fig. 2.2).
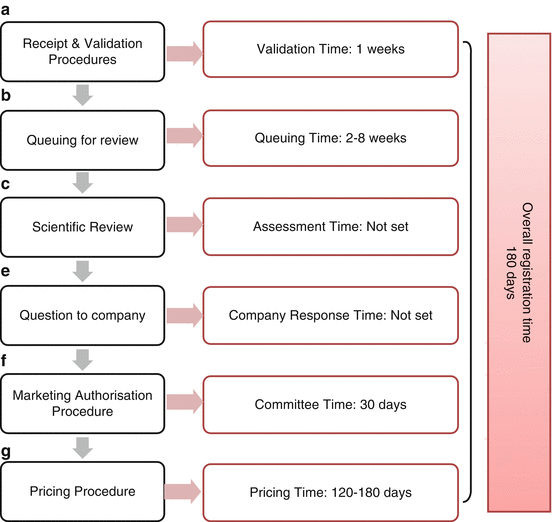
Fig. 2.2
The regulatory review process map for Kuwait
The most important goal of the Kuwaiti review process is to ensure that (a) the product is registered and marketed in countries with recognised and competent regulatory authorities for at least 12 months; (b) that the product meets the desired, internationally recognised, quality standards to ensure that the product was manufactured for its intended use; (c) that the product is stable for the entire proposed shelf life and for 6 months under the stressed conditions of 40 °C/75 % relative humidity; and (d) that the product price must be reasonable and affordable for local patients.
The Submission Phase
The review process starts with the local agent (or the sponsor) submitting the registration dossier along with a covering letter to the Director of Kuwait Drug and Food Control (KDFC) officially requesting the registration of the pharmaceutical product. The authority, then, transfers the registration dossier to the registration department, and the Drug Registration and Release Superintendent (DRRS) acknowledges the receipt and appoints a reviewer to undertake the assessment of the dossier. The product is placed in a queue for review by the department’s administrative staff member who is responsible for keeping a record of the dossiers to be transferred to the appointed reviewing staff member.
The Evaluation Phase
After entering the scientific review stage, the reviewer evaluates the chemical and manufacturing control (CMC) data focusing on the following data:
Product specifications and detailed methods of analysis of the finished products with the reference pharmacopoeias
Full-stability studies in tabulated form addressing the proposed product shelf life
Raw material specifications and their methods of analysis as well as the reference pharmacopoeia
Even though the authority does not evaluate safety and efficacy data, it considers documentation of such data as an important part of a successful approval process. Therefore, sponsors must ensure that safety and efficacy data are submitted to the authority along with all other registration documents. These are addressed when further investigations are necessary and then the following procedure occurs. The authority indicates that New Active Substances (NAS) companies must submit clinical studies as a major requirement for a successful approval of their NAS. Clinical studies are sent to the relevant specialised hospital or health institutions for evaluation by clinical experts, and a report is sent back to the regulatory authority stating the clinical effectiveness of the product on selected patient volunteers and whether there is a significant clinical need for such a medicine in Kuwait. The authority appends this report to the scientific assessment report. In the case of an EAS, the sponsor must submit bioequivalence studies to provide evidence of bioequivalence between the locally marketed NAS and its EAS counterpart under registration.
Kuwait requires suitable facilities, expertise, resources and proper settings to be able to conduct the desired standard safety and efficacy assessments. Therefore, the main focus of the authority is on the pharmaceutical quality data that provide the assurance that the drug was formulated for its intended use. Furthermore, administrative documents such as the certificate of pharmaceutical product (CPP), the list of countries where the product is registered and marketed with the registration dates, the good manufacturing practice (GMP) certificate and a manufacturing licence authenticated by the health authority in the country of origin are the official documents that are requested from the sponsor to overcome the shortage in resources and expert capacities to evaluate the safety and efficacy studies. For completion of the review process, NASs do not enter the quality control analysis stage as long as the sponsor has provided complete pharmaceutical quality documents to ensure that this product is of the desired quality. Once this is achieved, the NAS is ready for approval. EASs, however, are sent to the QC laboratory for sample analysis. The results must comply with analytical results and ranges provided by the manufacturer’s certificate of analysis of the finished product. Moreover, the results must not be outside the ranges and limits provided by the NAS company’s patent counterpart. Furthermore, to overcome the lack of post-marketing surveillance (PMS) capacity, the authorities request the necessary pharmacovigilance reports as part of the safety studies submitted along with the registration dossier.
After reviewing the dossier, questions are collected as they arise during the scientific assessment and sample analysis. These are sent to the sponsor after the Drug Registration and Release Superintendent (DRRS) has given their advice by signing the question/query form. The authority places no limit on the sponsors’ processing time and the scientific assessment clock stops at this point until a reply is received from the sponsor. This step affects the overall approval time when delays in the sponsor’s reply are encountered. However, the authority does not exclude it from the review process but considers its impact on the final approval time.
The Authorisation Phase
When the full assessment has been successfully completed, the final approval decision is made by the DRRS which is officially endorsed by the director of the authority. At this stage, the pricing negotiations have been completed and an agreement has been reached. Once the review and pricing procedures are completed, the product is finally approved and the sponsor is then required to pay the fees to receive the registration certificate. The agreed product price is listed in the next supplement to be presented for approval by the Health Minister. Once the Minister approves the price, it is officially published in the locally distributed business magazine called ‘Kuwait Today’, after which the product is ready to be marketed.
The key milestones in the approval process and the associated timelines in Kuwait are illustrated in Fig. 2.2. The target approval time in Kuwait is 120–180 days for both NASs and EASs. However, this timeline is not fully enforced due to many interfering factors that hinder its implementation such as the clock stop during the sponsor’s response time with no specific time limit for the sponsor to process the authority’s questions and queries. Nevertheless, if the sponsor does not respond to the authority’s question within a maximum period of 2 years and is still willing to complete the registration process in Kuwait, the original dossier is returned to the sponsor and a new application must be made.
Regulatory Review Process in Oman
A thorough evaluation of the regulatory process in Oman was undertaken and the milestones identified. The regulatory review process in Oman comprises ten stages, which are considered critical and have an impact on the approval time of medicines (Fig. 2.3).
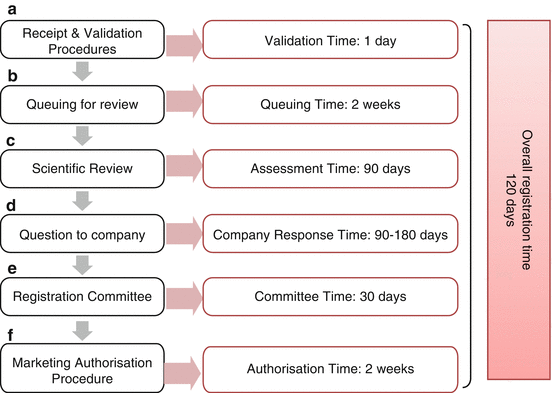
Fig. 2.3
The regulatory review process map for Oman
The Submission Phase
As a common practice, the sponsor submits the product registration file to the authority. All documents must be completed for official acceptance. The following items are checked at the validation stage:
Legal status of the applicant/local agent
GMP status of the manufacturer
Organisation of the registration dossier
Certificate of pharmaceutical product (CPP) authenticated by the respective embassy or consulate general
If the application is incomplete, the dossier is rejected and a new application must be made after providing the missing data. After receiving the product dossier, the company must pay the registration fees within 1 week. Once the validation stage is successfully completed, applications join the queue and have to wait for 2 weeks before being allocated for review. There is no official priority review procedure for fast-track medicines, but life-saving products are unofficially prioritised.
The Evaluation Stage
The product enters the scientific review stage, and data on quality, safety and efficacy are assessed in parallel. The safety and efficacy parts are reviewed in the drug control department and the quality part by the quality control laboratory department. There is a formal record for the starting time of the scientific assessment. In the primary scientific assessment procedure, an internal reviewer in the drug control department completes a scientific product report, detailing the trade, EAS names, indication and country of origin. Then, the product assessment report is sent to the scientific committee for evaluation. This committee assesses the product report and generates questions, queries and concerns relevant to the product’s quality, safety and efficacy.
The committee also examines any queries that are raised during the assessment process. These questions are returned to the reviewer to be collected in one batch for the sponsor after the scientific committee has given its advice. After sending the questions and queries to the sponsor, there is a time limit of 90–180 days given to sponsors to reply to the questions which are entirely dependent on the type of queries addressed, whether they are related to major or minor issues. The sponsor can meet with internal staff to discuss questions and queries that arise during the assessment, but they are only permitted to meet the directors and/or section heads.
The drug control department refers the marketing authorisation application assessment report and their recommendation to the registration committee within 90 days of its receipt, and the registration committee makes a decision within 30 days from the date of receipt.
The registration committee consists of members from two directorates in the Ministry of Health: (1) six members from the Directorate General of Pharmaceutical Affairs and Drug Control and (2) two members from the Directorate General of Medical Supply and all members are pharmacists. Meanwhile, the laboratory sample analysis is carried out in parallel with the scientific review, but the analytical step can be waived if the product is registered in Saudi Arabia, UAE and/or Kuwait or if it is registered in the GCC Central Drug Registration (GCC-DR).
The Authorisation Phase
Finally, the registration committee is responsible for granting the marketing authorisation and pricing of the product after completion of the review process. A product registration certificate is issued within 2 weeks after the committee has provided a positive decision about the product registration which is signed by the chairperson of the registration committee. If the registration committee rejects the application, the sponsor can appeal within 60 days from the date of receiving the committee’s decision; otherwise, a whole new submission is required after a 60-day period.
The key milestones in the approval process and the target approval time in Oman are illustrated in Fig. 2.3. The length of the scientific assessment, and, therefore, the approval time depends on the type of product being reviewed and the regulatory requirements to register such a product, whether they are major or minor requirements.
Regulatory Review Process in Qatar
An evaluation of the regulatory review process in Qatar was undertaken and the milestones were identified. The regulatory review process in Qatar is illustrated in Fig. 2.4 and consists of 13 critical steps that have an impact on the overall time of patient access to the medicines.
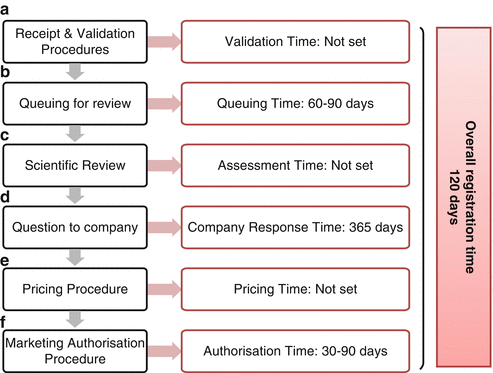
Fig. 2.4




The regulatory review process map for Qatar
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
