and FJE Vajda2
(1)
Clinical Neurology and Neuropharmacology, University of Queensland, and Honorary Consultant Neurologist, Royal Brisbane and Women’s Hospital, Brisbane, QLD, Australia
(2)
Department of Medicine and Neurology Director of the Australian Epilepsy and Pregnancy Register, University of Melbourne and Royal Melbourne Hospital, Melbourne, Australia
Abstract
This chapter outlines the pharmacokinetics and clinical pharmacology of the longer established antiepileptic drugs that continue to be reasonably often used in pregnancy, to provide a background to considering the alterations pregnancy and its aftermath produce in these parameters. The four drugs considered, viz. phenobarbitone, phenytoin, carbamazepine and valproate, are cleared from the body mainly by virtue of metabolism, and the clearance values of all increase progressively during pregnancy, though the change for carbamazepine is relatively small unless it is co-administered with another antiepileptic drug that induces the activity of drug-metabolising enzymes. The literature contains information regarding the individual metabolic pathways for phenytoin and carbamazepine biotransformation during pregnancy, indicating that the increased clearances of these drugs are due mainly to the development of increased capacities of already existing metabolic pathways rather than the appearance of new metabolic pathways. Relatively little information has been published concerning the dispositions of these drugs in the neonate, though there are data on their concentration ratios between maternal plasma and breast milk.
This chapter and the following two attempt to deal with the clinical pharmacology of the antiepileptic drugs for which useful pregnancy-related data are available in the current literature. In the accounts that follow, the main emphasis is given to the pharmacokinetics of the drugs, because pregnancy alters antiepileptic drug pharmacokinetics rather than other aspects of their pharmacology. At the end of the section, considering each individual drug in what it is hoped will be sufficient detail for background purposes, there is a separate portion concerning the effects of pregnancy on the drug’s pharmacokinetics. These portions considering the situation in pregnancy are referenced in some detail, whereas the referencing of the remainder of the material for each drug is less exhaustive, both to conserve space and because detailed contemporary accounts of the topic outside pregnancy are available (e.g. Levy et al. 2002; Engel and Pedley 2008; Shorvon et al. 2009). A few overall reviews of the pharmacokinetics of the antiepileptic drugs in pregnancy have been published (e.g. Pennell 2003; Tomson et al. 2013).
In Chaps. 4 (older drugs), 5 (newer drugs) and 6 (less commonly used drugs), the individual antiepileptic drugs are dealt with largely in the order of their first appearance in clinical use, though drugs that have had comparatively small usages in women of childbearing age, and those for which relatively little information relevant to pregnancy is available, are considered after the more widely used agents have been discussed.
It should be appreciated that the values of the pharmacokinetic parameters of a given drug in the non-pregnant state may remain relatively constant over long periods in adult life, whereas the values change throughout the course of pregnancy. Hence, because the published values may have been determined at different stages of pregnancy (with the stage sometimes not being specified), the parameter values for pregnant women tend to show wider ranges of variation than those that apply in the relatively unchanging non-pregnant state.
Certain pharmacokinetic parameters, in particular drug clearance, have sometimes been expressed in the literature relative to body weight, and sometimes not, and also over periods of either an hour or a day. In the following chapters, clearances have been expressed on a per hour basis in the interests of uniformity, but it has not been possible to express them consistently on, or not on, a body weight basis because of lack of the requisite weight data in some published studies.
Phenobarbitone
Phenobarbitone (phenobarbital ) is the oldest antiepileptic drug that remains in reasonably frequent present-day use. Its ability to prevent seizures was recognised as long ago as 1912. The drug’s use in Western countries is diminishing even though it is a reasonably effective agent. Because of its low cost, it remains one that is still widely employed in many other parts of the world. Over the years, a number of its molecular derivatives have been synthesised and used to treat epilepsies. The only derivative still employed, though now only on a modest scale, is primidone (desoxy-phenobarbitone ) which probably acts mainly through the phenobarbitone to which it is biotransformed. However, the primidone molecule possesses sedative actions in its own right and is believed to be useful in treating essential tremor . The other phenobarbitone congener that had some recent use was N-methylphenobarbitone, which has an interesting stereospecific pattern of metabolism. Only phenobarbitone itself is considered further in this chapter, with a few comments added concerning available primidone data.
Chemistry
Phenobarbitone (5-ethyl-5-phenyl barbituric acid) is usually provided for oral use as 30, 50 and 100 mg tablets. The corresponding more water-soluble sodium salt is usually employed in parental preparations of the drug. Phenobarbitone has a pKa value of 7.2 and a molecular weight of 232.23.
Pharmacodynamics
At a molecular level, the antiepileptic effects of phenobarbitone are thought to depend on its binding to specific receptors on the GABAA –chloride ion channel mechanism in neuronal cell membranes. The binding prolongs the opening of the ion channel and so hyperpolarises the cell membrane. Phenobarbitone ’s effects tend to be somewhat similar to those of GABA -ergic agents. At supratherapeutic doses the drug may also bind to voltage-dependent cell membrane Na+ ion channels .
Clinically, phenobarbitone is reasonably effective against all of the commonly encountered types of focal and primary generalised epileptic seizure types apart from absences.
Pharmacokinetics
Absorption
The available information suggests that orally administered phenobarbitone has a fairly rapid absorption rate. However peak plasma phenobarbitone levels may not occur for several hours after oral drug intake. This happens because the drug’s slow elimination results in the amount absorbed per unit time after each dose exceeding the amount being cleared from the body for a relatively long period. Unlike several other injectable antiepileptic drugs, phenobarbitone seems to be absorbed relatively quickly after intramuscular injection. No values for the oral bioavailability of the drug seem to be readily available, but clinical experience suggests that it is reasonably fully absorbed from the available preparations of the drug that are marketed for oral administration.
Distribution
Phenobarbitone’s apparent volume of distribution is around 0.5 L per kilogram, consistent with its being distributed throughout body water without there being any major accumulation in a particular body component. In infants and neonates, the value of the apparent volume of distribution seems to be higher. The plasma protein binding of the drug is lower in the neonate than in the adult, in whom about 50 % of the drug in plasma is protein bound. The binding percentage in plasma remains constant over the drug concentration range of 20–100 mg/L. The drug’s concentrations in cerebrospinal fluid , saliva and milk are largely similar to the simultaneous concentrations of the protein-unbound drug in plasma.
Elimination
At therapeutic concentrations, phenobarbitone appears to be eliminated by processes which follow linear kinetics. The elimination half-life in adults is 3–4 days, though it is shorter in children (mean 37 h as compared with 73 h in adults; Garrettson and Dayton 1970).
Guelen et al. (1975) cited a phenobarbitone oral clearance value of around 0.012 l per kilogram per hour in young children, falling to around 0.004 L per kilogram per hour by the age of 12–15 years and thereafter remaining reasonably constant throughout adult life.
Excretion Unchanged
The range of published values for the proportion of a phenobarbitone dose excreted unchanged in urine has been rather wide, the most reliable average figure being around 25 %, consistent with the drug being cleared mainly by metabolism.
Metabolism
Part of a phenobarbitone dose (perhaps some 30 %) follows a rather unusual metabolic pathway, being excreted in urine after conversion to an N-glucoside conjugate . This, together with unmetabolised drug, probably accounts for a little over half of a phenobarbitone dose under steady-state conditions. Much of the remainder of the dose is converted to various hydroxy derivatives (Fig. 4.1). None of phenobarbitone’s metabolites appear to possess pharmacological activity.
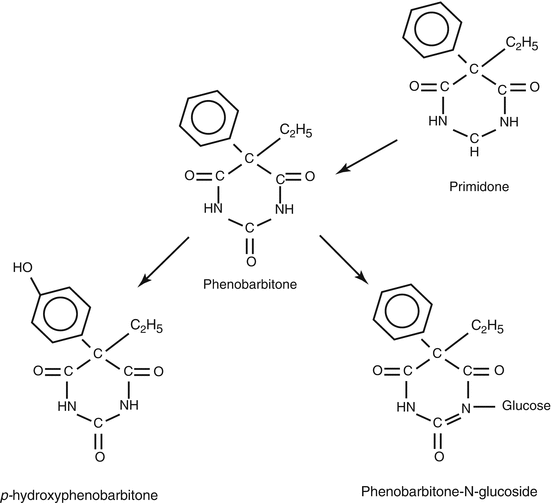
Fig. 4.1
The relationship between the molecular structures of phenobarbitone and primidone, and the two main phase I metabolites of phenobarbitone
Clinical Pharmacokinetics
Phenobarbitone at plasma levels around 10 mg/L has some effectiveness in controlling seizures in the types of epilepsy that respond to the drug. Above that threshold value, the proportion of patients whose seizures respond increases with increasing plasma drug concentrations. The upper level of the beneficial concentration of the drug varies from person to person. A representative, though perhaps conservative figure for the upper limit, might be 30 mg/L. Patsalos et al. (2008) cited a therapeutic range of 10–40 mg/L. Schmidt et al. (1986) found that a mean plasma phenobarbitone concentration of 18 mg/L was associated with full control of the tonic–clonic seizures of primary generalised epilepsies , but a mean plasma level of 37 mg/L was required before control of simple or complex partial seizures was achieved, whether or not these seizures became secondarily generalised.
Travers et al. (1972) reported that, relative to the phenobarbitone dose, females tended to have lower plasma levels of the drug than males, though the difference was not statistically significant. Eadie et al. (1976, 1977) noted no difference in the steady-state relationship between plasma phenobarbitone concentration and drug dose between 4-year-old and older males and females. However, compared with their male counterparts, younger females required substantially lower phenobarbitone doses to attain the same steady-state plasma phenobarbitone concentrations.
Interactions
Phenobarbitone is one of the classic inducers of the microsomal mono-oxygenase enzyme system that catalyses the oxidative metabolisms both of numerous endogenous substances and also exogenous molecules, including various therapeutic drugs (Gillette 1963; Conney 1967). Lecamwasam et al. (1975) showed that continuous administration of phenobarbitone to humans for more than 7 days caused increased formation of liver microsomal protein and heightened activities of various cytochrome P450 enzymes, including CYP3A4 which catalyses numerous drug biotransformations. As a result of this enzyme induction , continuing phenobarbitone intake is associated with decreased plasma concentrations of endogenous molecules such as bilirubin, cortisol, folate and unconjugated oestriol . Carbamazepine , ethinyl oestradiol , phenytoin , valproate and warfarin are among the more relevant drugs whose clearances are affected by the induction, but there are numerous others. As mentioned in Chap. 2, the decreased circulating levels of oestrogenic hormones resulting from concurrent administration of phenobarbitone may compromise the effectiveness of certain oral contraceptives that have relatively low oestrogen contents. The kinetics of vitamin K are not affected by phenobarbitone-mediated enzyme induction. More extensive accounts of the drug’s interactions are available in the reviews of Patsalos and Perucca (2003a, b).
Several authors have noticed that phenytoin intake tends to cause a rise in plasma phenobarbitone concentrations (Morselli et al. 1971; Lambie et al. 1976; Windorfer and Sauer 1977). In contrast, co-administered carbamazepine and clonazepam have little effect on circulating phenobarbitone levels. Simultaneous administration of valproate causes a notable increase in plasma phenobarbitone concentrations (Vakil et al. 1976; Wilder et al. 1978; Bruni et al. 1980). Kapetanovic et al. (1981) showed that the co-administration of valproate lengthened the half-life of phenobarbitone and decreased its clearance . This change is associated with falls in the urinary excretions of its oxidative metabolite p-hydroxy phenobarbitone and the drug’s N-glucoside conjugate (Bernus et al. 1994). Folic acid administration has been reported not to alter plasma phenobarbitone levels (Baylis et al. 1971) but there have been contrary findings (Mattson et al. 1973; Eadie et al. 1977).
Adverse Effects
Phenobarbitone has dose-related sedative effects which trouble some patients even at quite low plasma drug concentrations. Some tolerance to the sedation may develop if low phenobarbitone dosages are used at the outset and increased very gradually, recognising that the rather long half-life of the drug means that new steady-state conditions may not apply until more than 2 weeks after a dosage change. Different patients respond to the sedation from the drug in different ways. Some simply become rather depressed, miserable and relatively inert; others become irritable and aggressive as they struggle against the slowing of their mental processes produced by the drug. In those with epilepsy, Reynolds and Travers (1974) found a statistical correlation between increasing plasma phenobarbitone concentrations in the range that is usually considered to be associated with therapeutic benefit and psychomotor slowing and personality change. In frank overdosage with the drug, increasing drowsiness , nystagmus , double vision and ataxia of gait occur.
Long-continued phenobarbitone administration can be accompanied by reduced plasma folate concentrations and rarely by macrocytic anaemia (Chanarin et al. 1958; Davis and Woodliff 1971). Hypocalcaemia and decreased bone mineral density have been reported. Critchley et al. (1976) found a 56 % incidence of Dupuytren’s contracture in the hands of chronic epileptic patients treated with phenobarbitone in a residential centre, but there do not seem to have been subsequent reports of this effect. Intake of the drug may precipitate attacks of porphyria in those predisposed to that disorder. Other complications such as skin rashes , agranulocytosis , aplastic anaemia and hepatitis are considerable rarities.
Pregnancy
The Mother
As mentioned in Chap. 3, in pregnant women Lander et al. (1977) noted that there was a changing relationship between phenobarbitone oral dosage and steady-state plasma phenobarbitone concentrations that was consistent with the apparent clearance of the drug increasing as pregnancy progressed. Employing the term ‘apparent clearance’ is necessary in this connexion, because in this study no proof was provided that the full oral dosage of the drug had been absorbed. In the previous year, Mygind et al. (1976) and Dam et al. (1976) had found no increase in the drug’s clearance in pregnancy, but during the following decade, Bardy et al. (1982), Hosokawa et al. (1984), Kan et al. (1984) and Yerby et al. (1990) all found that the behaviour of the phenobarbitone dose to plasma concentration relationship in pregnancy was consistent with the drug’s clearance being increased, particularly in the later stages of pregnancy. There seems to have been little subsequent exploration of the matter, but it now seems fairly generally accepted that phenobarbitone’s clearance usually increases in pregnancy and returns to its baseline value after childbirth. Increased biotransformation probably accounts for the increased clearance, but studies demonstrating this do not seem to have been reported. It would be interesting to know what happens to the capacity of the drug’s unusual N-glucosidation pathway during pregnancy.
Battino et al. (1984) noted a tendency for plasma primidone levels to rise, relative to drug dose, in the second trimester of pregnancy, coinciding with a decrease in plasma phenobarbitone concentrations derived from the drug. There appears to be no further information available regarding the mechanisms involved in the change. With the declining use of primidone, it seems unlikely that an explanation will become available.
The Foetus
In ex vivo studies in perfused human placenta cotyledons obtained shortly after delivery, Kluck et al. (1988) found that phenobarbitone was comparatively rapidly transferred across the placenta to the foetal compartment. They obtained no evidence that the placenta could metabolise the drug. De Carolis et al. (1992) reported that, in five mother–infant pairs at the time of birth, plasma phenobarbitone concentrations in maternal and umbilical cord plasma were very similar. Earlier, Melchior et al. (1967) and Ishizaki et al. (1981) had both shown that phenobarbitone concentrations in the neonate’s umbilical cord plasma were some 95 % of the simultaneous concentrations in maternal plasma.
Breast Milk
Coradello (1973) could not detect measurable phenobarbitone concentrations in the milk of lactating women who were taking the drug. However, in a series of studies, Kaneko et al. (1979, 1982, 1984) found that the mean milk to maternal plasma phenobarbitone concentration ratios were, respectively, 45.9, 36.1 and 34.6 %. Davanzo et al. (2013) cited a figure of 40–60 % for the parameter.
The Neonate
Phenobarbitone has been used to control seizures in the neonate and some information is available regarding its pharmacokinetic properties in that age group. The drug’s apparent volume of distribution appears to be proportionately greater than in the adult. Painter et al. (1977) quoted a value of 0.97 ± 0.20 L per kg for the parameter, Pitlick et al. (1978) a very similar value and Fischer et al. (1981) a mean value of 0.81 L per kg. In neonates, some 57–64 % of the drug in plasma is not protein bound (Bossi 1982). Boreus et al. (1975) observed that the elimination half-life of phenobarbitone in neonates was inversely related to the plasma phenobarbitone concentration. Pitlick et al. (1978) found that the elimination half-life shortened from a mean of 115 h at the end of the first neonatal week to a mean of 67 h by the end of the fourth week. Fischer et al. (1981) cited a half-life value of 103.4 h and Ishizaki et al. (1981) a mean half-life value of 74.0 h. The parameter values cited earlier in this paragraph apply to the initial dose of phenobarbitone received by neonates not previously exposed to the drug. No information is available concerning the metabolic pathways involved in eliminating the drug in this age group. It is unclear whether the elimination rates would be similar in neonates whose mothers had taken the drug throughout pregnancy, in which case the neonate’s drug-metabolising capacity may have already been induced before birth.
Phenytoin
Phenytoin was developed as an antiepileptic agent in the late 1930s as the outcome of an attempt to discover new chemicals with molecular structural resemblances to phenobarbitone and with similar or greater antiepileptic efficacies. The drug remains in widespread use after more than two-thirds of a century, though in more affluent societies it is less often employed than formerly.
Chemistry
Phenytoin (5,5′-diphenylhydantoin ), a white crystalline material (pK a ~ 8.4), is marketed for oral use as the free acid (molecular weight 252.3) or as the sodium salt (molecular weight 274.3). The latter is more water soluble, though still poorly so. Oral suspensions of the drug, and a few solid dosage forms, contain the drug as the free acid. Most oral dosage forms contain the sodium salt. Sodium phenytoin preparations contain about 8 % less active substance than preparations containing the same weight of drug but in the form of the corresponding free acid. The solution of the sodium salt for intravenous or intramuscular injection has a pH of around 12.
Pharmacodynamics
In the past, numerous possible biochemical mechanisms were proposed to explain the antiseizure action of phenytoin, but it now appears clear that, at therapeutically relevant concentrations, the drug’s antiepileptic action is mediated through its effects on cell membrane voltage-dependent Na+ ion channels (Mantegazza et al. 2010). Phenytoin binds to, and then prolongs the inactivation state of, Na+ channels (mainly α-subtypes 1.1, 1.2, 1.3 and 1.6 – Qiao et al. 2014). This is particularly the case when the channels are in their fast-inactivated state (Karoly et al. 2010). The drug’s effect is greater when the cell membrane is already depolarised and in an inactive state than when the membrane is hyperpolarised (Thomas and Petrou 2013). The Na+ channel block becomes use dependent if the cell membrane is depolarised repeatedly. Phenytoin ’s inactivation of voltage- and frequency-dependent Na+ channels renders partly depolarised axons less able to transmit rapid trains of action potentials (as occurs in epileptic discharges) but has less effect on relatively infrequent and more physiological action potential traffic along axons. The drug binds to the same site in the inner pore of sodium channel as carbamazepine and lamotrigine do (Lipkin and Fozzard 2010).
At high concentrations, phenytoin may inhibit axonal and axon terminal calcium channels , thereby potentially stabilising the cell membranes and decreasing the release of excitatory neurotransmitters, in particular glutamate , from axon terminals which have been activated by the arrival of action potentials. It also increases chloride conductance at GABAA receptors . This, at least in theory, might have an antiseizure effect. Phenytoin has no action at the T-type calcium channels in the thalamus that are involved in the genesis of absence seizures (Kuo 1998).
Phenytoin ’s rather selective inhibition of fast action potential traffic along axons seems to account for its ability to impede the spread of epileptic activity while having relatively little effect on more physiological rates of transit of neuronal axon traffic. It also helps explain the observation that the drug tends not to prevent seizure discharge initiation but deters discharges spreading.
Clinically, phenytoin is useful in controlling most forms of epileptic seizure disorder, though not the absence seizures of idiopathic, i.e. genetic, generalised epilepsy . The drug’s action on cell membrane Na+ channels has also been utilised to treat cardiac arrhythmias, myotonia and certain other disorders.
Pharmacokinetics
The clinical pharmacokinetics of phenytoin was reasonably fully worked out a generation ago. Little additional knowledge has subsequently become available.
Absorption
The available information concerning the oral bioavailability of phenytoin was reviewed by Neuvonen (1979). The occurrence of a formulation-related bioavailability issue in Australasia nearly 50 years ago should have alerted the medical community to the drug’s potential bioavailability issues, but reports of generic phenytoin tablets whose oral bioavailabilities appear incomplete and also possibly inconsistent continue to appear in the literature (e.g. Berg et al. 2008). Storage of the drug under conditions of high temperature and humidity may impair its bioavailability.
Intramuscular administration of phenytoin is too inefficient to be useful clinically, because the drug is absorbed very slowly and inconsistently from its administration site. Fosphenytoin , a water-soluble phenytoin prodrug, is better absorbed after intramuscular administration (Fischer et al. 2003). Intravenously injected phenytoin is fully bioavailable, but the solution is highly alkaline and contains polyethylene glycol. Consequently, it must be administered very slowly to minimise unwanted effects. Furthermore, the drug may crystallise from solution if injected into an intravenous fluid reservoir containing a solution at a more physiological pH, e.g. glucose saline.
Distribution
In humans, phenytoin is distributed throughout total body water. Published values for the drug’s apparent volume of distribution have been in the range 0.5–0.8 L/kg. There seems to be little or no selective regional concentration of the drug. Its concentrations in plasma are a little higher than those in red blood cells. Brain phenytoin levels are slightly higher than simultaneous drug concentration in plasma. Under steady-state conditions, the drug’s concentration is higher in cerebral white than grey matter. Phenytoin is transported out of the brain by a P-glycoprotein mechanism situated in cells in the blood–brain barrier. Animal studies have shown that, if this extrusion mechanism is well developed or is induced following repeated seizures, brain phenytoin concentrations become disproportionately low relative to simultaneous plasma water drug concentrations. This effect may be associated with apparent treatment resistance in experimental animals with seizures (Loscher 2007). It is not yet clear whether a similar situation applies in human epilepsy (French 2013).
About 90 % of the phenytoin in adult plasma is bound to plasma proteins, mainly albumin. The protein-unbound fraction of the drug in plasma is higher in neonates than adults and also increases a little with advanced age, in late pregnancy, in the presence of hypoalbuminaemia from various causes and in that of high glycated albumin levels, as occur in diabetics. Phenytoi n concentrations in cerebrospinal fluid and routinely collected saliva , tears and sweat are very similar to the unbound concentrations of the drug in plasma. However saliva phenytoin concentrations vary with saliva flow rate and are affected by the presence of gum disease (Kamali and Thomas 1994).
Elimination
Phenytoin is unusual among drugs in present-day therapeutic use in that, at clinically relevant dosages, it exhibits Michaelis–Menten rather than linear elimination kinetics. Nevertheless, the linear kinetic parameters of elimination half-life and clearance are often cited for the drug. The numerical values of the latter parameters are not constant in a given individual but vary with the phenytoin concentration range over which the values have been determined. The calculated half-life is longer and the clearance lower, at higher plasma drug concentrations than at lower ones in the same person. A commonly quoted value for the drug’s half-life over the concentration range likely to be encountered in human therapeutics is 22 ± S.D. 9 h, with a clearance value for adults of 0.02 L/kg/h, and for children below 5 years of age, one of 0.06 L/kg/h.
Most published values for the Michaelis constant (K m) of phenytoin have ranged between 3 and 30 mg/L, with the mean around 6 mg/L (24 μmol). This is a lower concentration than the conventional lower limit of the therapeutic range of plasma concentrations of the drug (10 mg/L). The saturable elimination of the drug produces a non-linear relationship between phenytoin dosage and steady-state plasma phenytoin concentration. Relatively small dose increases are associated with disproportionately large increases in plasma phenytoin concentrations. Published values for the maximum velocity of phenytoin elimination (V max) have been in the range 6–16 mg/kg/day [17.5–46.7 mg/h]. The values are higher on a body weight basis in young children than in adults (Bauer and Blouin 1983).
Excretion Unchanged
Less than 5 % of a phenytoin dose is excreted in urine unchanged, the drug being eliminated mainly through metabolism.
Metabolism
The main known metabolite of the drug, accounting for 60–80 % of the dose, is the pharmacologically inactive oxidation product p-hydroxy phenytoin (5-phenyl, 5′-p-hydroxyphenylhydantoin, HPPH). The conversion of the parent molecule to p-hydroxy phenytoin, via a postulated short-lived arene oxide intermediate, is catalysed by the CYP450 isoforms CYP2C9 and CYP 2C19 and, in the skin, possibly by CYP2C18. Much of the p-hydroxy phenytoin formed in the phase I biotransformation stage appears in urine as a glucuronide conjugate formed in a reaction catalysed by UDP glucuronosyl transferase 1A isoforms (Nakajima et al. 2002). Other quantitatively minor oxidation products are known, e.g. phenolic, dihydrodiol, catechol and O-methyl catechol derivatives (Fig. 4.2). The liver is responsible for nearly all of the biotransformation of the drug, but some may occur in the gums, skin and other peripheral tissues.
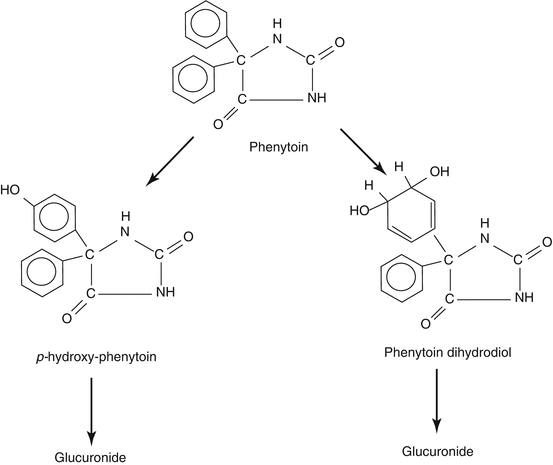
Fig. 4.2
Main pathways of phenytoin metabolism. The heavier arrows indicate the pathways whose capacity is increased in pregnancy
At conventional dosages in humans, CYP2C9 activity is responsible for some 90 % of the hydroxylation of phenytoin, the [S]-isomer of p-hydroxy phenytoin being formed preferentially. The [R]-isomer of p-hydroxy phenytoin is formed via activity of CYP2C19 . Poor metabolisers of the now superseded hydantoin derivative mephenytoin (methoin) form relatively little [R]-p-hydroxy phenytoin (Ieiri et al. 1995). CYP2C9 polymorphisms may account for the occasional individuals in Western populations and the more frequent ones of Asian extraction, who cannot tolerate conventional phenytoin dosages (Kesavan et al. 2010).
Clinical Pharmacokinetics
Kutt et al. (1964) nominated a range of plasma phenytoin concentration between 10 and 20 mg/L (40–80 μmol) as being usually associated with the best chance of achieving seizure control without producing drug overdosage manifestations. This concentration range has come to be regarded as the ‘therapeutic’ or ‘target’ range for the drug. However, other authors have proposed different lower and upper limits of the range, e.g. from 7 mg/L (28 μmol) to 25 mg/L (100 μmol) (Loiseau et al. 1977). In some patients, seizures are controlled at plasma concentrations above or below the 10–20 mg/L range, without unwanted effects of the drug occurring (Patsalos et al. 2008). Schmidt et al. (1986) found that a mean plasma phenytoin concentration of 14 mg/L (56 μmol) sufficed to control generalised tonic–clonic seizures, but a mean concentration of 23 mg/L (92 μmol) was required to control partial seizures . The therapeutic range values for the drug in plasma water (i.e. the unbound plasma concentration) and in saliva are approximately one-tenth of those that apply for whole plasma.
For plasma phenytoin concentrations in the ‘therapeutic’ or ‘target’ range, new steady-state conditions should apply 4–8 days after a phenytoin dosage change. At higher circulating drug concentrations steady-state conditions may be more delayed after a dosage change (up to about 2 weeks). Under steady-state condition, the peak-to-trough fluctuation in therapeutic range plasma phenytoin concentration in adults is likely to be of the order of ± 10 % over a 12 h dosage interval. There are greater inter-dosage fluctuations over the same time interval in children.
In a population taking commonly used adult dosages of phenytoin (300 or 400 mg/day), there is a wide range of steady-state plasma phenytoin concentrations. An appreciable proportion of the values falls outside the therapeutic range . Expressing drug dosage relative to body weight improves the correlation. In the average adult, a phenytoin dose of 5 mg/kg/day will be associated with a mean mid-therapeutic range steady-state plasma phenytoin concentration of 15 mg/L (60 μmol). Prepubertal children, with their higher velocities of elimination of the drug, are likely to require double that dosage to achieve a similar mean steady-state plasma drug concentration. Neonates and children in the first few months of life and the elderly require slightly lower doses on a body weight basis than the average adult.
As mentioned above, if a patient’s phenytoin dosage has to be increased, it is important to realise that the Michaelis–Menten elimination kinetics of the drug will cause plasma levels of the drug to increase more than if linear elimination kinetics had been applied.
If the presence of a reduced plasma protein-binding capacity for phenytoin is suspected, measurement of unbound plasma phenytoin or salivary phenytoin concentrations is likely to provide a more reliable basis for interpreting the clinical situation than the whole plasma drug concentration value.
Some workers have reported that, for a give phenytoin dose, adult females have statistically significantly lower plasma phenytoin levels than adult males (Sherwin et al. 1974; Richens 1975). Others have not detected such differences (Eadie et al. 1973; DeLeacy et al. 1979). The latter workers noted that when oral contraceptives were being taken, plasma phenytoin concentrations tended to be higher relative to drug dose than in women not taking these agents.
In some women, plasma phenytoin levels tend to fall, relative to the drug dose, around the time of menstruation (Rościszewska et al. 1986). Shavit et al. (1984) showed that the clearance of phenytoin tended to be higher and the elimination half-life shorter, at the time of menstruation as compared with the values of these parameters at the mid-interval of the menstrual cycle .
Interactions
Over the years, numerous interactions between phenytoin and endogenous substances and co-administered drugs have been described (Nation et al. 1990; Patsalos and Perucca 2003a, b; Patsalos 2013). Nearly all the interactions are pharmacokinetic in nature and mainly involve altered rates of metabolism of one or both of the substances involved. The interactions that have been described are too numerous for detailed individual consideration here.
Interactions Altering Phenytoin Concentrations
A few interactions involve altered phenytoin absorption, but these are unlikely to prove an issue during pregnancy. Certain endogenous substances and various acidic drugs (e.g. salicylates , valproic acid , heparin ) may displace phenytoin from its plasma protein-binding sites. These interactions are also unlikely to cause subsequent difficulties during pregnancy unless free (i.e. protein-unbound) drug concentrations are used as a guide to the adequacy of antiepileptic therapy, as may be done particularly in late pregnancy.
Interactions that alter phenytoin metabolism are a different matter. Taking drugs which inhibit phenytoin metabolism may cause raised phenytoin concentrations and possibly overdosage manifestations. For example, co-administration of antiepileptic drugs such as ethosuximide , felbamate , oxcarbazepine and sulthiame may result in increased plasma phenytoin levels relative to the drug dose. Interactions which involve induction of phenytoin metabolism, thus lowering its plasma concentrations, are more common. Such interactions may go unnoticed, unless plasma phenytoin concentrations are monitored or seizure control deteriorates. Primidone , vigabatrin , carbamazepine and phenobarbitone may be responsible for such interactions. However, the opposite effect sometimes occurs when carbamazepine or phenobarbitone is added to phenytoin therapy. The effects of valproic acid on plasma phenytoin levels appear to be inconsistent.
Phenytoin Affecting Other Substances
Phenytoin may alter the elimination of other substances by inducing: (1) the synthesis of the CYP isoenzymes responsible for their metabolism, including not only CYP2C isoforms but also CYP3A4 (which catalyses many drug oxidations) and CYP1A12, and (2) the synthesis of certain glucuronyl transferases involved in drug conjugation. As well, co-administered phenytoin may inhibit the metabolism of other CYP2C9 or CYP2C19 eliminated drugs. Drugs that may be encountered in pregnant women, and whose plasma concentrations are likely to be reduced in the presence of phenytoin include, among antiepileptic agents, carbamazepine , clobazam , clonazepam , felbamate , lamotrigine , primidone , tiagabine , topiramate , valproic acid , ethosuximide and zonisamide . Phenytoin ’s effect on circulating phenobarbitone concentrations is variable. Plasma levels and clinical effectiveness of many cardiovascular agents, chemotherapeutic agents, hormonal agents, psychotropic agents and other substances may be reduced by concurrent phenytoin administration.
The references cited at the beginning of the present chapter provide additional information regarding the drug’s numerous described interactions.
Adverse Effects
Numerous adverse effects of phenytoin have been described over its long period of extensive use in humans. Some adverse effects are clearly dose related; others probably involve hypersensitivity reactions, possibly related to immune-mediated processes.
Nervous System
Phenytoin overdosage tends to produce manifestations of vestibulocerebellar disturbance before sedation becomes troublesome. There may be horizontal nystagmus , typically at plasma phenytoin concentrations above 20 mg/L (80 μmol). At concentrations above 30 mg/L (120 μmol), ataxia of gait and double vision occur. At concentrations above 40 mg/L (160 μmol), drowsiness , sometimes with nausea and vomiting, and, at still higher concentrations, coma develop (Kutt et al. 1964). There are considerable interindividual differences in the correlation between phenytoin concentration and adverse effects. Some patients experience unwanted effects even at plasma levels below the conventional therapeutic range . Other patients appear untroubled at plasma phenytoin levels above 30 mg/L (120 μmol). Occasional patients may experience mood disorders, mainly depression, as the dose of the drug is increased. Paradoxically, seizure control may deteriorate in some patients as plasma phenytoin levels become ‘supratherapeutic’ or if the drug has been prescribed for epilepsy syndromes that involve absence or myoclonic seizures . Rarely, phenytoin overdosage results in various dyskinetic and dystonic involuntary movements, asterixis or ophthalmoplegia. Shorvon and Reynolds (1982) reported that the drug may cause subclinical, and occasionally clinically recognisable, peripheral neuropathy .
Skin and Gums
Within the first few days of phenytoin intake, 5–10 % of patients given the drug develop a measles-like rash , usually appearing first on the trunk. If phenytoin intake is not ceased promptly, more extensive and serious skin and internal organ involvement can develop. Other cutaneous reactions, e.g. Stevens–Johnson syndrome , systemic lupus erythematosus , exfoliative dermatitis and toxic epidermal necrolysis, are less frequent. Continued phenytoin intake may cause an overgrowth of body hair , particularly in dark-haired women. Acne and coarsening of facial features can develop.
Intravenous administration of phenytoin may lead to a ‘purple glove’ syndrome with progressive skin discoloration, oedema and pain which involve the hand and forearm distal to the administration site (O’Brien et al. 1998). Too rapid intravenous infusion of the drug is better avoided. As well, thrombophlebitis may develop in the vein into which the drug has been administered. The incidence of this adverse effect appears to be lower when intravenous fosphenytoin is used (Fischer et al. 2003).
Gum hyperplasia may develop in between 13 and 40 % of those taking phenytoin . The topic has been reviewed on several occasions (Meraw and Sheridan 1998; Ayra and Gulati 2012). The severity of the gum hyperplasia seems related to the plasma phenytoin level, though poor dental hygiene makes the hypertrophy more obvious.
Bone
Long-term phenytoin intake, alone or together with another older antiepileptic drug, can be responsible for the development of reduced bone mineral density and sometimes overt osteomalacia resulting from increased bone turnover. Plasma calcium levels may fall and alkaline phosphatase levels increase, with reduced plasma 25-hydroxycholecalciferol concentrations. The problem is likely to be worse if the diet is poor in vitamin D content and if there is little exposure to sunlight. Induction of vitamin D metabolism by the drug, and possibly impaired intestinal absorption of dietary calcium, appears to be responsible.
Lymphoid Tissue
Rarely, a widespread but reversible lymphadenopathy , a pseudolymphoma syndrome , can develop after long-term phenytoin use. Histologically the appearance of the affected lymph glands resembles that of Hodgkin’s disease. Even more rarely, true lymphoma has been reported in association with the drug.
Folates
Continued phenytoin intake may cause reduced plasma and red blood cell folate levels, the extent of the reduction being related to the plasma phenytoin concentration. The mechanisms involved are not fully elucidated. In patients taking long-term phenytoin therapy, the folate deficiency may occasionally result in megaloblastic anaemia .
Cardiovascular Effects
Oral phenytoin therapy in usual dosages is very unlikely to cause cardiovascular disturbances. Intravenous administration of the drug is potentially hazardous, there being dangers of hypotension, cardiovascular collapse and central nervous system depression.
Other Effects
Phenytoin intake can precipitate attacks of porphyria in sufferers from the disorder. If paroxysmal hypoglycaemic symptoms from an insulinoma are misdiagnosed as manifestations of epilepsy and treated with phenytoin, the real diagnosis may be further delayed because phenytoin can diminish pancreatic insulin secretion. As a result, the drug for a time may appear to provide successful therapy. Rare adverse effects of the drug include hepatitis , vasculitis, interstitial lung infiltration, interstitial nephritis , myopathy , thyroiditis , arthritis and the suppression of the formation of particular lines of blood cell.
Phenytoin intake can produce various asymptomatic biochemical effects. These include raised plasma levels of γ-glutamyl transpeptidase, alkaline phosphatase, high-density lipoprotein (HDL) cholesterol, caeruloplasmin, copper, prolactin and sex hormone-binding globulin. It may also cause reduced plasma concentrations of folate (discussed above), IgA, IgG, IgE, IgM, fibrinogen , thyroxine, triiodothyronine (but not free T4 and T3), protein-bound iodine, vitamin K , vitamin E, vitamin D metabolites (mentioned above), cortisol, oestrogens, progesterone , free testosterone , pyridoxal phosphate, tryptophan and thiamine. As mentioned in Chap. 2, the reduced circulating steroidal sex hormone levels may be associated with a diminished libido and with other disturbances of sexual functioning.
Pregnancy
The Mother
Once the altered relationship between phenytoin dose and plasma phenytoin concentrations during pregnancy was recognised in the 1976–1977 period (see Chap. 3), the explanation for the changes was sought. One possibility was that the oral bioavailability of the drug was decreased during pregnancy. Ramsay et al. (1978) described a single instance in which this may have occurred. In the 22nd week of her pregnancy, the woman involved appeared to excrete 56 % of her daily phenytoin dose (of 900 mg) unchanged in faeces. At 3 months and subsequently at 30 months postpartum, by which time the oral dose of the drug had fallen to 400 mg a day, the proportion of the dose lost in faeces had decreased, initially to 40 % and finally to 23 % of the oral dose. No further instances of such altered phenytoin absorption from the alimentary tract during pregnancy have been reported. Lander et al. (1984) carried out an opportunistic formal bioavailability investigation in five women who suffered their first epileptic seizures during pregnancy. Before commencing regular phenytoin intake, each woman received an initial intravenous dose of phenytoin followed a week later by an oral dose of the same magnitude. Except for folate , which all the women were taking, none of the women took any other agent known to affect plasma phenytoin concentrations. The calculated oral bioavailability fraction for the drug ranged from 0.79 to 1.01, with a mean value of 0.91 ± 0.10 (Fig. 4.3). It therefore appears unlikely that an incomplete oral bioavailability is the usual explanation for the drug’s altered dose to steady-state concentration relationship during pregnancy.