Chapter 24 The Metabolism of Membrane Lipids
Biological membranes contain phosphoglycerides, sphingolipids, and cholesterol (see Chapter 12). All of these membrane lipids can be synthesized in the body, and most are made in the cells in which they are used. However, considerable quantities are transported in the blood as constituents of plasma lipoproteins. This chapter discusses the biosynthesis and degradation of the membrane lipids.
Phosphatidic Acid Is an Intermediate in Phosphoglyceride Synthesis
Phosphoglycerides are synthesized in the cytoplasm and endoplasmic reticulum (ER) of all cells. Phosphatidic acid, which is synthesized from glycerol phosphate, is the key intermediate. Its biosynthesis is shown in Figure 24.1.
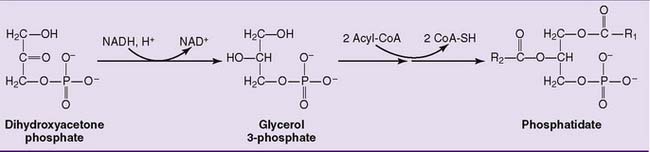
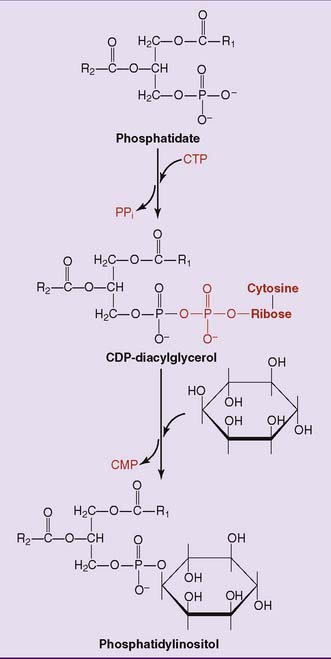
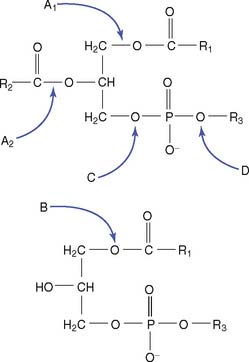
De novo synthesis of phosphoglycerides occurs via two pathways. In the phosphatidic acid pathway, phosphatidate is activated as cytidine diphosphate (CDP)-diacylglycerol. This strategy is used for the synthesis of phosphatidylinositol (Fig. 24.2) and cardiolipin.
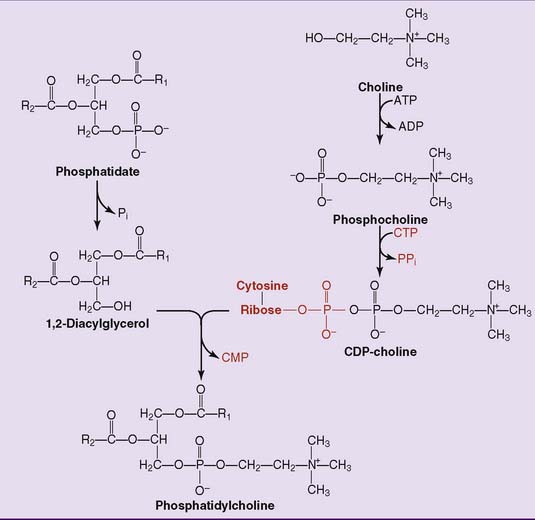
In the salvage pathway, phosphate is removed from phosphatidate to form 1,2-diacylglycerol. As shown in Figure 24.3 for the synthesis of phosphatidylcholine, choline is activated as the CDP derivative, and phosphocholine is then attached to the 3-hydroxyl group of 1,2-diacylglycerol. Phosphatidylethanolamine is synthesized in a similar way. Use of CDP-activated precursors for the synthesis of membrane lipids is analogous to use of uridine diphosphate–activated precursors for the synthesis of glycogen and other complex carbohydrates. However, in the case of the phosphoglycerides, one of the two phosphates in the CDP derivative remains in the product.
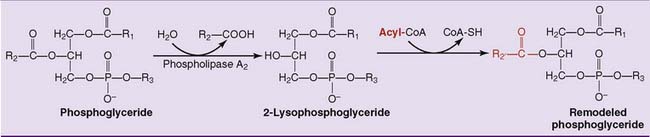
Phosphoglycerides Are Remodeled Continuously
Phospholipases are used to remodel the phosphoglycerides by changing the fatty acids in positions 1 and 2 (Fig. 24.4). They are named according to their cleavage specificity:
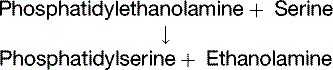
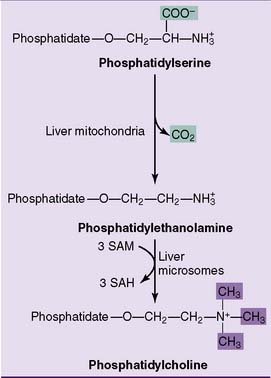
Phosphatidylethanolamine can be made by decarboxylation of phosphatidylserine, whereas phosphatidylcholine can be formed by methylation of phosphatidylethanolamine (Fig. 24.5).
Plasmalogens, which constitute up to 10% of the phosphoglycerides in muscle and brain, have an unsaturated fatty alcohol instead of a fatty acid in position 1 of the glycerol. Their synthesis is shown in Figure 24.6.
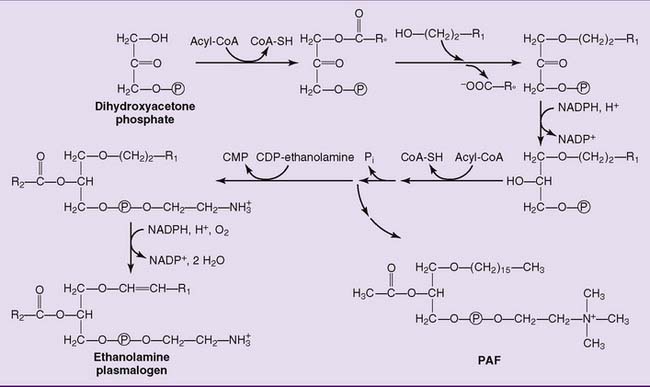
Platelet-activating factor (PAF) (see Fig. 24.6) is formed by white blood cells as a mediator of hypersensitivity reactions and acute inflammation. In concentrations as low as 10−11 to 10−10 mol/L, it induces platelet adhesion, vasodilation, and chemotaxis of polymorphonuclear leukocytes. The presence of an acetyl group in position 2, instead of a long-chain acyl group, makes PAF sufficiently water soluble to diffuse through an aqueous medium.
Sphingolipids Are Synthesized from Ceramide
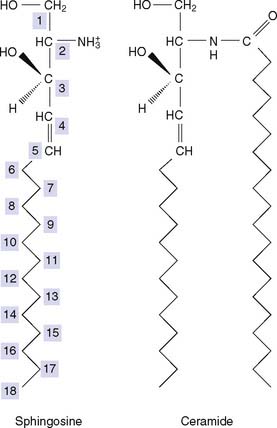
The primary hydroxyl group at C-1 of the sphingosine moiety carries either phosphocholine (in sphingomyelin) or carbohydrate (in the glycosphingolipids) (see Fig. 12.5). Sphingosine is synthesized in the ER of most cells from palmitoyl-CoA and serine, and the fatty acid is introduced from its CoA-thioester. The fatty acid usually is saturated and can be very long, with 22 or 24 carbons.
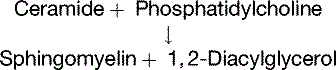
Deficiencies of Sphingolipid-Degrading Enzymes Cause Lipid Storage Diseases
Sphingolipids are degraded in the lysosomes. The breakdown of complex glycosphingolipids proceeds by stepwise removal of sugars from the end of the oligosaccharide (Fig. 24.8). Each of these enzymes is specific for the monosaccharide that it removes and the type of glycosidic bond that it cleaves.
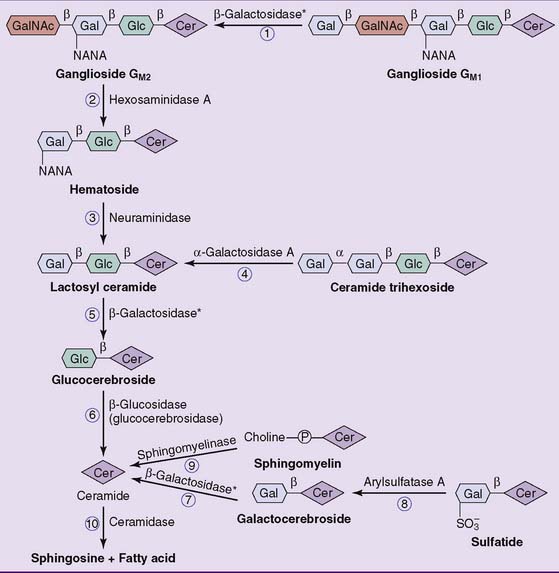
Figure 24.8 Lysosomal degradation of sphingolipids. The numbered reactions refer to the storage diseases listed in Table 24.1. There are two different β-galactosidases, one for ganglioside GM1 (reaction 1) and the other for galactocerebroside (reaction  ). Lactosyl ceramide (Cer) is degraded by both (reaction
). Lactosyl ceramide (Cer) is degraded by both (reaction  ). Gal, Galactose; GalNAc, N-acetylgalactosamine; Glc, glucose; NANA, N-acetylneuraminic acid.
). Gal, Galactose; GalNAc, N-acetylgalactosamine; Glc, glucose; NANA, N-acetylneuraminic acid.
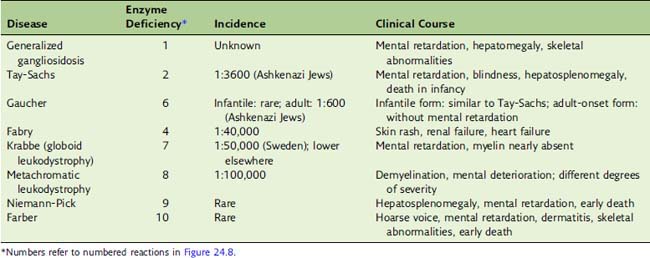
A deficiency of any of these enzymes leads to the accumulation of its substrate in the lysosomes. The resulting disease is called a lipid storage disease or sphingolipidosis (Table 24.1). The enzyme deficiency is expressed in all tissues. The nervous system is seriously affected in essentially all cases because of its high sphingolipid content and turnover. Hepatosplenomegaly is another common finding in these diseases because phagocytic cells in spleen and liver remove erythrocytes from the circulation, and nondegradable lipid from the red blood cell membrane accumulates in these tissues.
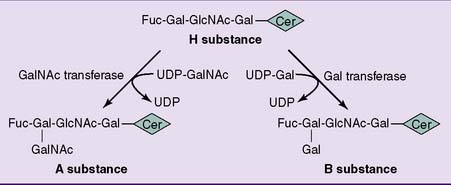
CLINICAL EXAMPLE 24.1: ABO Incompatibility
The antigens of the ABO blood group system are glycosphingolipids whose antigenic specificities are caused by variations in the terminal sugar of the oligosaccharide (Fig. 24.7). The glycosyl transferase that adds the last monosaccharide comes in three alleles. The A allele codes for an enzyme that adds N-acetylgalactosamine, the B allele codes for an enzyme that adds galactose, and the O allele has a nonsense mutation and produces no enzyme.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


