the portal vein, and hepatic artery and lymphatics. The functional unit of the liver is the hepatic acinus (Figure 15-2). The hepatic acinus is a three-dimensional structure with the portal tract as the central point (zone 1) where blood flows from terminal branches of the portal vein and hepatic arteries into the sinusoids and empties into the terminal hepatic venules at the periphery of the acinus (centrilobular/zone 3). Bile is secreted into the canaliculi and flows toward the portal areas into the interlobular bile ducts that are connected to the canaliculi by canals of Hering. The acinus thus includes parts of several adjacent lobules.
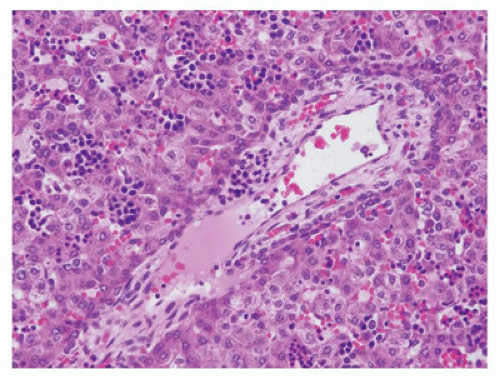 FIGURE 15-1 • Ductal plate in the fetal liver is formed by a collar of epithelial cells at the periphery of the portal tract and abuts against zone 1 hepatocytes. Note the presence of extramedullary hematopoiesis (H&E stain, 200×). |
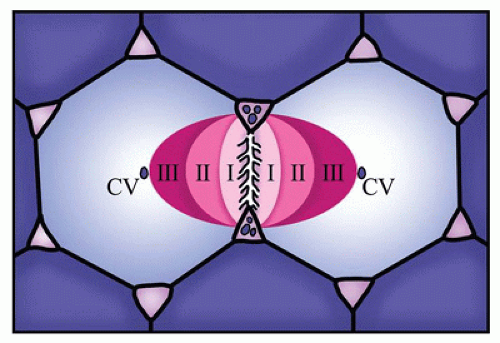 FIGURE 15-2 • Schematic view of hepatic lobule or acinus. The conventional view of the liver consisted of a hepatic lobule with a central vein (CV) surrounded by hepatocyte cords radiating to peripheral portal areas. The functional unit of the liver, the acinus, however, consists of a three-dimensional structure with a central portal tract surrounded by concentric zones of hepatocytes (I, II, and III), with the most peripheral zone (III) lying near the central vein. |
the right hemidiaphragm. In cases of omphalocele, the liver is often herniated into the omphalocele sac. In large omphaloceles, there is often distortion of the liver and its vascular and biliary connections. The liver may have signs of marked congestion and even hemorrhagic necrosis. Intrapericardial herniation of the liver occurs rarely and may result in massive pericardial effusion in neonates. Nearly all cases of the thoracopagus type of conjoined twins show connections between the two livers, ranging from a bridge to a common liver between the two twins.
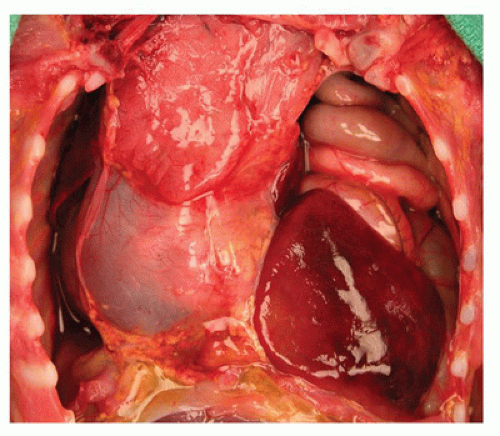 FIGURE 15-3 • Herniation of the liver through diaphragmatic defect. A large defect in the left leaflet of the diaphragm has led to the herniation of the left lobe of the liver and intestines into the left hemithorax, resulting in mediastinal shift to the right and severe pulmonary hypoplasia. |
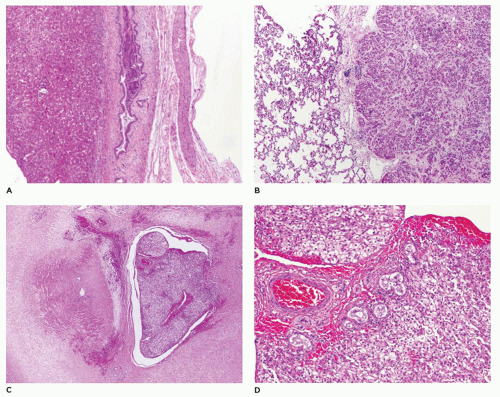 FIGURE 15-4 • A: Ectopic liver tissue within the diaphragm B: Lung C: Umbilical cord (H&E, 40×) D: Ectopic liver tissue in the umbilical cord with bile ducts (H&E stain, 200×). |
with glycogen storage diseases (GSDs) to maintain optimal preservation of glycogen. With formalin fixation, up to 70% of glycogen is lost due to the soluble nature of the predominant form of glycogen in the cytoplasm. Glycogen can be preserved with freezing and/or alcohol fixation, allowing for quantitative evaluation by analytical techniques (frozen tissue) and qualitative assessment by histochemical staining (periodic acid-Schiff [PAS], PAS diastase). Quantitative analysis of enzyme(s) responsible for suspected metabolic and mitochondrial diseases must be done on frozen tissue. Assessment of metals such as copper and iron may require use of metal-free containers to freeze liver tissue for proper quantification. Assessment of gene mutation and sequencing of the gene responsible for the enzyme defect or mitochondrial disease also requires frozen tissue. Preservation of the enzyme, enzyme activity, DNA, and RNA requires cryopreservation at -70°C and maintaining this temperature until the tissue reaches the appropriate reference laboratory. Depending on the testing required for a definitive diagnosis, tissue requirements may dictate an open biopsy of the liver and/or skeletal muscle. Obtaining fibroblast cultures from a skin biopsy may also be necessary for genetic and enzyme studies. The current trend in surgical and interventional radiology practice has been toward needle core biopsies for diagnosis. The pathologist should be aware of necessary tissue requirements (tissue weights and preservation methods) for appropriate testing to be completed. A single tissue core of 20 mm in length from a 16-gauge needle with a 1.5 mm diameter yields about 15 mg of tissue. Several metabolic disease tests require a minimum of 20 mg of tissue. With GSDs, 100 mg or more of tissue will be needed. This may necessitate numerous tissue cores or an open biopsy to obtain adequate tissue for all tests. This emphasizes the importance of active communication between the health care team and the pathologist. Because tissue will be preserved in a steady state with cryopreservation (-70°C), comprehensive workup (histopathology, histochemistry, electron microscopy) by the pathologist to determine which additional testing is most appropriate can be completed prior to performing specialized testing on the frozen tissue.
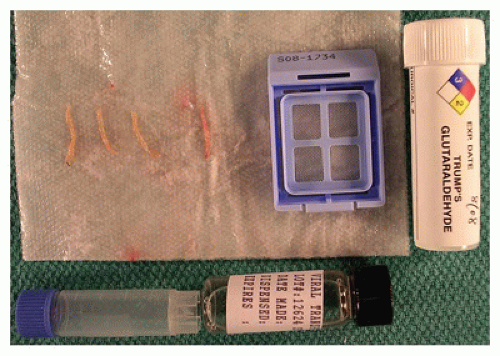 FIGURE 15-5 • Liver biopsy triaging consists of tissue submitted for formalin fixation and paraffin embedding, freezing tissue at -70°C, viral or microbiologic culture submission, and glutaraldehyde fixation for electron microscopy. |
(9,10). This syndrome has a mutation in the ABCC2 gene that is responsible for synthesis of MRP2/cMOAT, an organic ion transporter. Zone 3 hepatocytes contain deposits of a granular golden-brown pigment, with staining characteristics of melanin. Loss of cMOAT staining is demonstrated by immunohistochemistry (IHC), a valuable tool in diagnosis of early cases when pigment deposition may not be evident. Ultrastructurally, however, these granules do not have the features of melanosomes, but are lysosomes with a distinctive appearance.
TABLE 15-1 JAUNDICE IN INFANTS DUE TO UNCONJUGATED HYPERBILIRUBINEMIA | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
may show ballooning, acidophilic necrosis including necrosis of giant cells, and pseudoglandular or acinar formation. Lobular or portal mononuclear cells are generally sparse, but a prominent inflammatory component and extramedullary erythropoiesis should suggest an infectious etiology. The portal areas in INH are usually not expanded, except for the sometimes prominent myelopoiesis, and the bile ducts are normal or may be inconspicuous. Rarely, there may be mild proliferation of the interlobular bile ducts, better demonstrated with a cytokeratin stain. Histologic features comparing INH with those of extrahepatic biliary atresia (EHBA) are listed in Table 15-3.
TABLE 15-2 CHOLESTATIC DISEASE IN INFANCY | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
and hydronephrosis) and group 3 is associated with laterality defects with cardiovascular, gastrointestinal, and splenic anomalies. Similar to neonatal hepatitis, EHBA is also considered to be a condition with more than one etiology. In fact, INH and EHBA have been seen as sequential processes in the same infant over a period of several weeks to months (17). While viral theories have been proposed as etiologies for EHBA, recent documentation of a high incidence of autoimmune diseases in first-degree relatives of all BA groups raises the possibility of an autoimmune mechanism (16). Defects in cilia have also been implicated in the pathogenesis of EHBA (18).
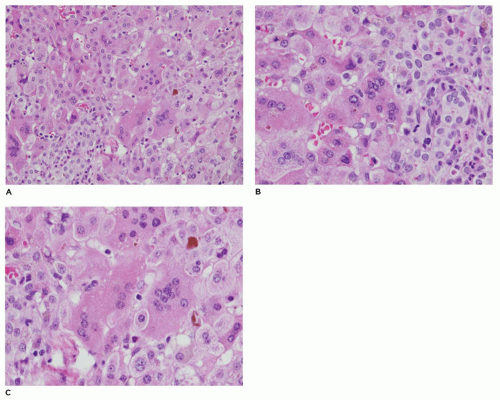 FIGURE 15-6 • A-C: Idiopathic neonatal hepatitis. Giant cell transformation with expansion of the portal region by chronic inflammatory cells, prominent bile ducts, and readily identified cholestasis (A at 100×, B at 200×, C at 200×, H&E). |
TABLE 15-3 COMPARISON OF BILIARY ATRESIA AND NEONATAL HEPATITIS SYNDROME | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
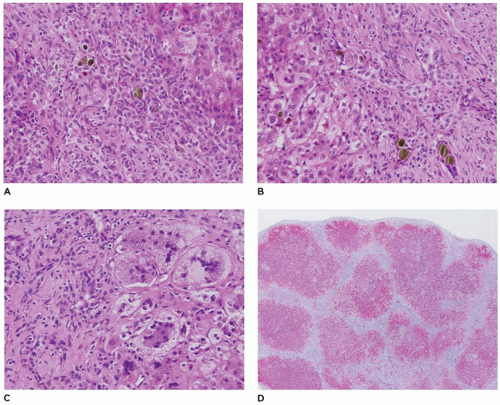 FIGURE 15-7 • A: EHBA liver biopsy. Histopathologic features include diffuse bile duct proliferation in expanded portal region with canalicular cholestasis. B: Hepatocytes organized into pseudoacinar pattern C: Giant cell transformation adjacent to fibrotic portal region D: Cirrhosis may occur rapidly without surgical intervention (H&E stains, A, B, C at 200×, D 40×). |
rosettes. Bile “lakes” consisting of amorphous collections of bile surrounded by inflammatory cells and connective tissue are seen rarely in liver biopsies, unlike in adults with obstruction of the biliary tract. Hepatocytes may display mild enlargement and rarefaction of the cytoplasm (feathery degeneration), but fatty change is rarely seen. Giant cell transformation, if present, is generally restricted to zone 1 at the interface with the expanded portal tracts (Table 15-3). Instances of hepatocyte and giant cell necrosis may be encountered.
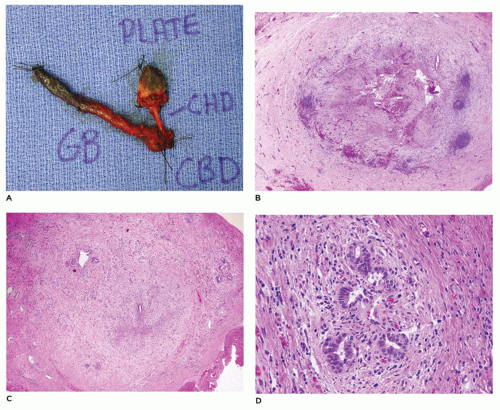 FIGURE 15-8 • Resection of residual common bile duct during Kasai procedure. A: Common bile duct remnant with orientation by surgeon. CHD, hepatic duct, GB; gallbladder, CBD; common bile duct; plate, liver plate. B: Near-total obliteration of common bile duct lumen with no residual epithelial lining (H&E, 20×). C: Microscopic residual common bile duct lumen with epithelial lining (H&E, 20×). D: Nests of bile duct epithelium in common bile duct wall (H&E, 200×). The latter side chain structures should not be mistaken as evidence of a patent bile duct. |
destruction, the stroma around and between ducts becomes infiltrated by neutrophils, lymphocytes, and macrophages, along with a prominent fibroblastic proliferation. As the ductular inflammation diminishes and the ducts are destroyed, the stromal activity is replaced by dense fibrosis, containing a few residual inflammatory cells and remnants of bile ducts. Rarely, islands of hyaline cartilage may be found in the porta hepatis, suggesting a congenital malformation as the cause of the atresia in these selected cases. The gallbladder may be diminutive and exhibit varying degrees of fibrosis, epithelial degeneration and destruction, and luminal compromise.
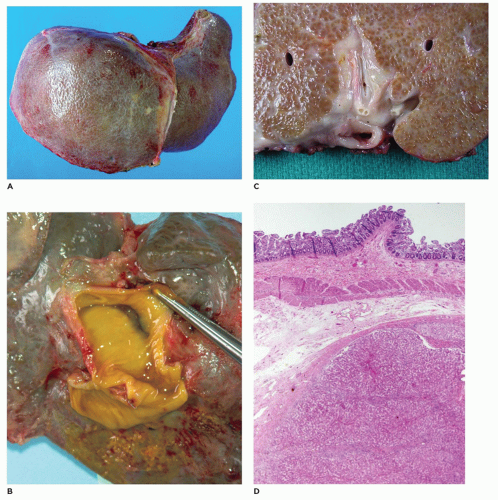 FIGURE 15-9 • Explanted liver with prior Kasai procedure. A: Explanted liver with micronodular cirrhosis. B: Patent small-bowel anastomosis site at liver hilum. C: Liver in cross-section with close apposition of small-bowel anastomosis to liver hilum and micronodular liver parenchyma with diffuse bile staining. D: Small-bowel anastomosis separated by muscular wall of small bowel and fibrous tissue from the underlying liver parenchyma (H&E, 10×). |
Absence of any lumen lined by biliary epithelium, with little or no inflammatory cells in the connective tissue (Figure 15-8).
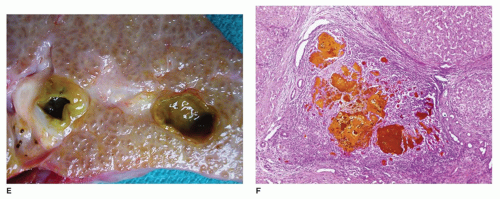
FIGURE 15-9 • (continued) E: Large bile-filled lakes within the liver parenchyma and micronodular cirrhosis with diffuse bile staining. F: Bile plugs distending portal region with adjacent micronodular liver parenchyma (H&E, 40×).
Presence of lumina lined by cuboidal epithelium. The remnants may be numerous, have lumens less than 50 µm, and are surrounded by a neutrophilic infiltrate. Cellular debris and bile may be present in the lumen, and epithelial necrosis may be seen in ducts with a diameter exceeding 300 µm.
The presence of a central altered bile duct incompletely lined by columnar epithelium, in addition to smaller epithelial structures resembling those in the second type.
approximately 50% of patients in some series, with approximately a 75% survival rate (29).
TABLE 15-4 FAMILIAL CHOLESTATIC SYNDROMES | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
familial cholestasis, which may explain the variable presentations and manifestations of the disease (33). There is still a group of familial cholestasis in which the exact genetic defect is still not known, and recent attempts have identified TJP2 and claudin genes as possible candidates (34). PFIC1 disease is associated with extrahepatic disease manifested by diarrhea, pancreatitis, and hearing loss, while BSEP disease (PFIC2) causes progressive liver failure and increased risk of malignancy with no extrahepatic disease (35,36,37). Gallstones may be seen in some of the BRIC2 patients (36).
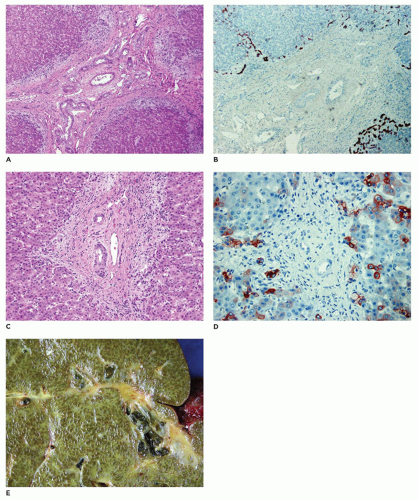 FIGURE 15-10 • A-D: Alagille syndrome (paucity of interlobular bile ducts). Absence of bile ducts within the portal tracts and presence of proliferating cholangioles at the periphery of the liver lobules as identified with cytokeratin 7 immunostaining (H&E staining, A, C: 20×; cytokeratin 7 immunostaining, B, D: 20×). E: Micronodular cirrhosis with bile plugs and diffuse bile staining. |
children with familial hypercholesterolemia have bland intracanalicular cholestasis and low GGT and improve with ursodeoxycholic acid treatment. The genetic defects in these children are associated with aberrant tight junction proteins (claudin, TJP2 gene) and a defective bile acid conjugation enzyme (gene BAAT) (42). More recent molecular methods have helped elucidate genetic defects in TJP2 gene in a larger subset of infants with progressive cholestatic liver disease who have low GGT and neither the ATP8B1 nor ABCB11 mutations (34).
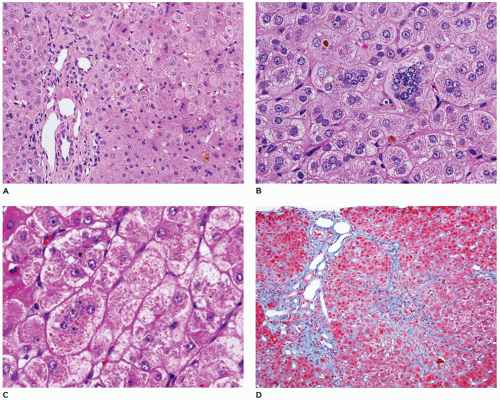 FIGURE 15-11 • A, B: Progressive familial intrahepatic cholestasis (PFIC). Hepatic lobular disarray with giant cell transformation and focal canalicular cholestasis (H&E, A: 100×, B: 400×). C: Diffuse cytoplasmic cholestasis of hepatocytes with granular bile (H&E, C 400×). D: Central lobular fibrosis with fine feathery extension into the peripheral zone toward the portal region (Trichrome, D: 100×). |
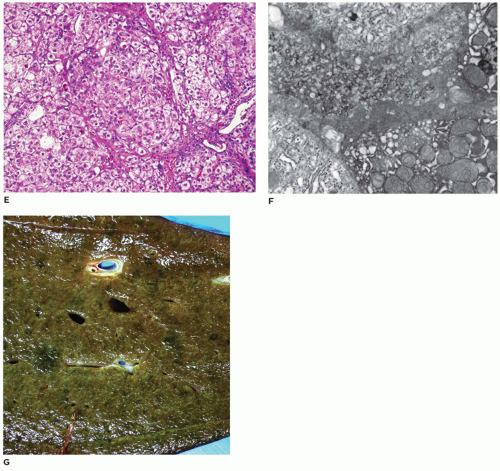 FIGURE 15-11 • (continued) E: Micronodular cirrhosis with portal to portal bridging fibrosis and loss of central veins (H&E, E, 100×). F: Electron microscopic appearance of coarse granular bile markedly distending a canalicular space between hepatocytes (electron microscopy, F: 20,000×). G: Gross appearance. |
Ultrastructural evidence of bile duct destruction in nonsyndromic paucity of bile ducts has been regarded as representing a primary ductal injury.
TABLE 15-5 CAUSES OF DUCTOPENIA | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
identify dilatation of the bile ducts, suggesting choledochal cyst or extrahepatic biliary obstruction.
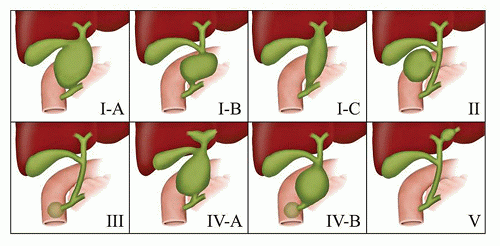 FIGURE 15-12 • Classification of congenital bile duct cysts. |
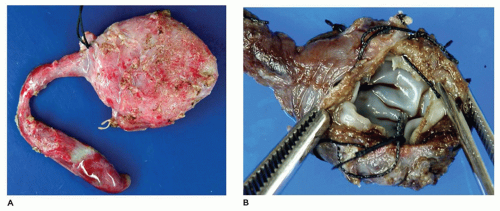 FIGURE 15-13 • Choledochal cyst. A: Choledochal cyst with portion of common bile duct. B: Choledochal cyst with smooth glistening lining of cystic cavity. |
renal lesions (Figure 15-14; Table 15-6) (55,56) including ARPKD (mutation of PKHD1—fibrocystin at 6p21 locus), ADPKD (mutation of PKD1—polycystin-1 or mutated PKD2—polycystin-2), and rarely in Meckel-Gruber syndrome (mutation of MKS1 at 17q, MKS2 at 1q, or MKS3 at 8q) and Jeune syndrome. The causative gene for ARPKD is PKDH1, which encodes for fibrocystin/polyductin that is located on the primary cilium together with other proteins that are defective in other renal cystic disease syndromes. Together, these constitute the group of diseases known as “ciliopathies.” Because Caroli disease and choledochal cyst are associated with CHF in a small proportion of cases, a common pathogenesis is worth consideration. CHF has also been seen in association with a variety of other syndromes including Joubert syndrome (agenesis or hypoplasia of the cerebellar vermis, retinal dystrophy, chorioretinal colobomata, oculomotor abnormalities, episodic hyperpnea, ataxia, neurodevelopmental delay), Ivemark syndrome (renal, pancreatic, hepatic dysplasia), Down syndrome, Laurence-Moon-Biedl syndrome (mental retardation, retinitis pigmentosa, obesity), and COACH syndrome (hypoplasia of the cerebellar vermis, oligophrenia, congenital ataxia, coloboma, hepatic fibrosis, (MKS3, CC2D2A, and RPGRIP1L genes) (55,57) (see Chapter 12).
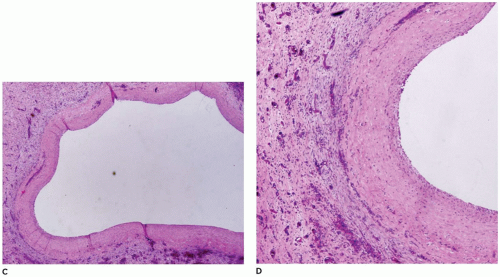 FIGURE 15-13 • (continued) C, D: Lumen of choledochal cyst lacking an epithelial lining with the wall formed by dense connective tissue with scattered chronic inflammatory cells and no residual smooth muscle (H&E, C: 20×, D: 40×). |
tendency for progression of the hepatic manifestations over long periods of clinical follow-up.
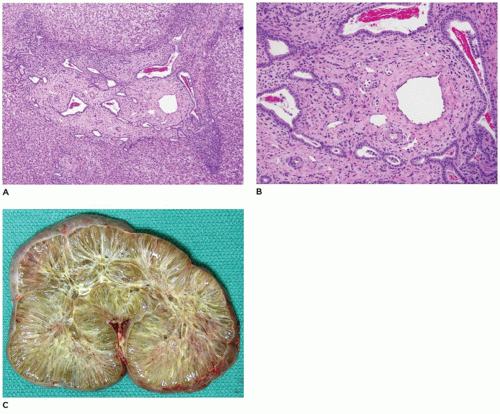 FIGURE 15-14 • CHF associated with autosomal recessive polycystic kidney disease. A, B: Ductal plate abnormality with dilated and concentric arrangement of bile ducts in expanded and fibrotic portal regions (H&E, A: 20×, B: 40×). C: Autosomal recessive polycystic kidney disease with markedly enlarged kidney due to numerous thin cysts extending from the cortical to medullary regions. |
disease are the features of this familial condition. Progressive renal failure occurs during the first two decades of life (62).
TABLE 15-6 CYSTIC LESIONS OF THE LIVER AND ASSOCIATED CONDITIONS | |
|---|---|
|
TABLE 15-7 DISTINCTIVE FEATURES IN METABOLIC DISORDERS WITH NEONATAL HEPATITIS-LIKE CHANGES | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
liver and other tissues (69,70). The GSDs most frequently associated with hepatic manifestations include types I, II, III, IV, VI, and IX. The mode of inheritance is autosomal recessive in all the types described, with the exception of type IXb, which is inherited as a sex-linked recessive trait. Some forms of the disease present in infancy and others in early childhood, with failure to thrive, developmental delays, acidosis, hypoglycemia, and hepatomegaly. The morphologic changes have been reviewed by McAdams et al. (69), and the ultrastructural features have been illustrated by Phillips et al. (71). Features of those glycogenoses that significantly involve the liver are summarized in Table 15-8.
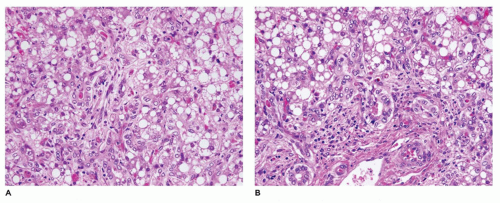 FIGURE 15-15 • Galactosemia. A, B: Hepatocytes display medium to large droplet fatty metamorphosis, along with pseudoglandular transformation (H&E, A: 100×, B: 200×). |
TABLE 15-8 HEPATIC FINDINGS IN GLYCOGEN STORAGE DISEASE | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
frozen sections or alcohol-fixed tissue. Lipid is also present, while fibrosis is typically absent. Rarely, Mallory bodies may be seen. Ultrastructurally, there are pools of monoparticulate glycogen in the cytoplasm and nuclei of hepatocytes. Lipid vacuoles are found in the cytoplasm. The organelles are displaced, and the size of the mitochondria may be increased. Hepatic adenomas have been reported with some frequency, and cases of HCC and hepatoblastoma have been described in type 1 glycogenosis (73,74). Hepatic transplantation has been used in the treatment of type I, III, and IV GSDs (75).
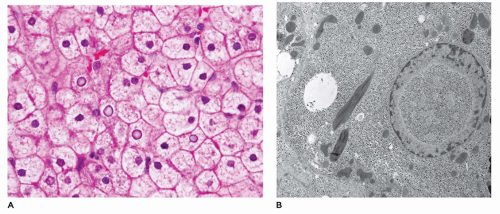 FIGURE 15-16 • Glycogen storage disease, type I. A: Hepatocytes distended by glycogen with obliteration of the sinusoids, forming a mosaic pattern, and glycogenated nuclei. (H&E, 400×.) B: Hepatocyte with glycogenated nuclei, cytoplasmic monoparticulate glycogen, large lipid droplet, and abnormally shaped mitochondria (electron microscopy, 7500×). |
but are digestible with pectinase treatment (Figure 15-18). Colloidal iron staining is also seen in the cytoplasmic inclusions. Ultrastructurally, the inclusions consist of central fibrillar glycogen surrounded by polyparticulate glycogen rosettes (69). Rare instances of diffuse reticuloendothelial involvement have also been reported (80). Prenatal diagnosis is possible.
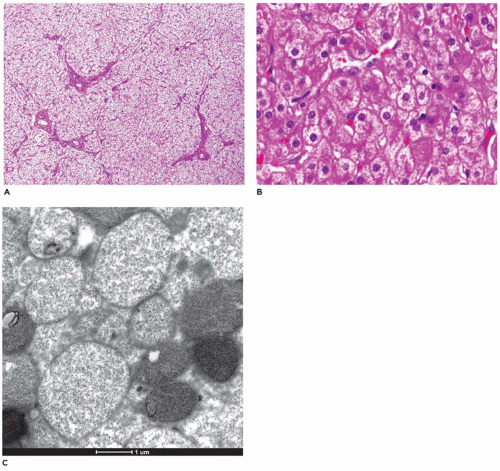 FIGURE 15-17 • Glycogen storage disease, type II. A: Hepatocytes demonstrating mosaic pattern with obliteration of sinusoids (H&E, 40×). B: Hepatocytes in close proximity to one another, with thickened cell membranes, fine cytoplasmic vacuolization, and indistinct sinusoids (H&E, 400×). C: Monoparticulate glycogen within membrane-bound lysosomes (electron microscopy, 20,000×). |
17q23-24) (72,81). Hepatic glycogenosis with stunted growth (type XI, Fanconi-Bickel syndrome) is associated with renal glycogen deposition. Type XI glycogenosis is caused by mutations in the glucose transporter 2 (GLUT2, 3q26.1-26.3) gene (82). Generalized glycogen deposition is accompanied by cirrhosis, but with normal glycogen metabolism.
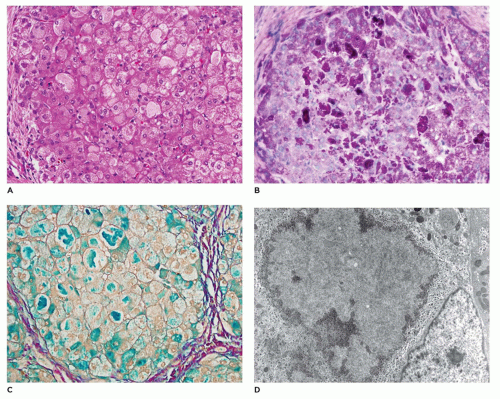 FIGURE 15-18 • A: Glycogen storage disease, type IV. Hepatocytes with pale hyaline inclusions surrounded by indistinct halos, resembling Lafora bodies (H&E, 200×). B, C: The inclusions stain intensely with PAS (400×) and colloidal iron (400×). D:Hepatocyte with hyaline inclusion comprised of fibrillary glycogen (electron microscopy, 6000×). |
Regenerative nodules may already be present in early liver biopsies. The chronic form is characterized by cirrhosis with variable-sized nodules separated by thick bands of fibrous connective tissue with little inflammation or bile duct proliferation. Hepatocytes may demonstrate nuclear hyperchromasia, dysplasia, or adenomatous hyperplasia. The incidence of HCC is quite high with tyrosinemia. Liver transplantation is advisable soon after diagnosis and before 2 years of age, because of the high risk of HCC (85,86). Treatment with NTBC [2(-nitro-4-trifluoromethylbenzoyl)-1-3-cyclohexanedione] prevents the accumulation of the toxic metabolites of FAH and the subsequent liver and neurologic effects, but does not entirely eliminate the risk of HCC (87).
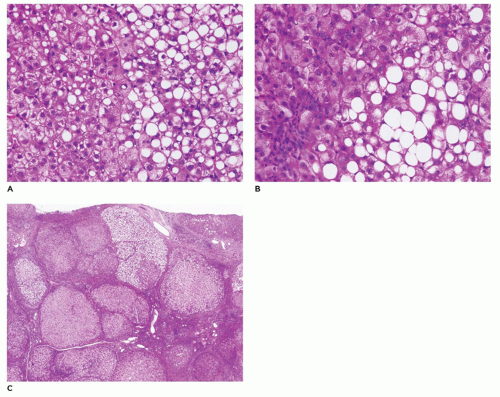 FIGURE 15-19 • Tyrosinemia. A, B: Hepatocytes with macrovesicular steatosis and indistinct sinusoid spaces (H&E, A: 200×, B: 400×). C: Micronodular cirrhosis in chronic form of tyrosinemia (H&E, 40×). |
at 19p13.3) (90,91). Many of the clinical stigmata of mucopolysaccharidoses may be seen, but mucopolysaccharides are not excreted in the urine. I-cell disease and pseudo-Hurler polydystrophy are autosomal recessive disorders. Coarse facies, skeletal changes, hepatosplenomegaly, and delayed growth and development are some of the clinical features. The primary histopathologic and ultrastructural changes are cytoplasmic vacuolization of hepatocytes, Kupffer cells, and, less frequently, biliary epithelial cells. Inclusions within clear vacuoles can be demonstrated within fibroblasts and peripheral nerves in skin and conjunctival biopsies (71). Glomeruli and renal tubular epithelium contain similar inclusions, and the inclusions are also present in the urine.
TABLE 15-9 LYSOSOMAL DISORDERS | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
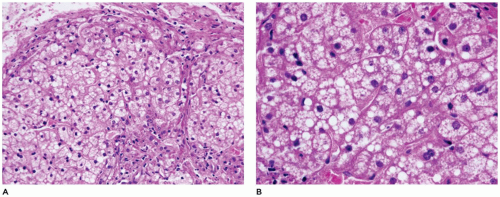 FIGURE 15-20 • A, B: Cholesterol ester storage disease. Hepatocytes with diffuse microsteatosis (H&E, A 200×, B 400×). |
19cen-q12), fucosidosis (FUCA1 gene at 1p34), and aspartylglycosaminuria (aspartylglucosaminidase gene at 4q32-33) (93). The liver is involved in all forms and has enlarged vacuolated hepatocytes. Ultrastructurally, the foamy appearance is due to cytoplasmic membrane-bound clear vacuoles (71). The vacuoles are of variable sizes, may be molded by adjacent vacuoles, and fuse to form larger vacuoles. They are composed of finely granular to flocculent material intermingled with membrane material. Kupffer cells, biliary epithelial cells, and endothelial cells show similar vacuoles.
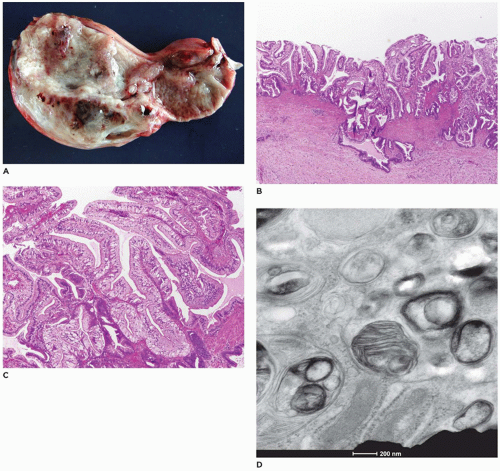 FIGURE 15-21 • Metachromatic leukodystrophy. A: Gallbladder with markedly thickened mucosa with fine cobblestone to papillary surface. B, C: Papillary fronds lined by columnar epithelial cells with amphophilic cytoplasm (H&E, B: 100×, C: 200×). D: Lysosomal inclusions with closely packed herringbone appearance (electron microscopy, 25,000×). |
(N-acylsphingosine amidohydrolase gene at 8p22-21.3) (95). Disseminated lipogranulomata are the morphologic findings. The liver is mildly affected, with clear vacuoles in the hepatocytes similar to the membrane-bound vacuoles in mucopolysaccharidoses. The Kupffer cells and portal macrophages have lysosomal comma-shaped, banana-shaped, and curvilinear inclusions in common with other tissues. Death occurs in adolescence or early adulthood (96).
 FIGURE 15-22 • Fabry disease. Membrane-bound lysosomal inclusions with lamellar and concentric pattern (electron microscopy, 12,000×). |
esterification (type II or type C, NPC gene at 18q11-12) (70,93). This disease is characterized by sphingomyelin storage in various organs. Sphingomyelin accumulation varies in extent, but it is most pronounced in type A (type 1A), the acute neuropathic form, and in type B (type 1B), the chronic nonneuropathic form. Sea blue histiocytes are seen in the bone marrow. The liver is enlarged and pale on gross examination. The lobular structure of the liver is not disorganized, and fibrosis is generally not a feature. However, cirrhosis may rarely occur.
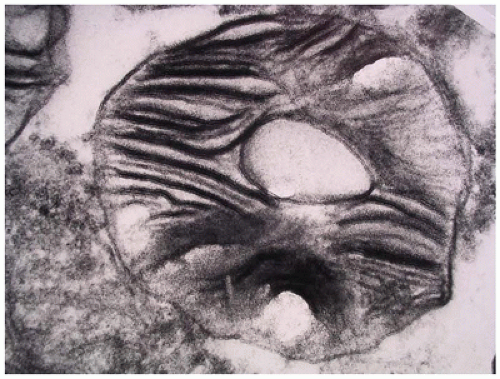 FIGURE 15-23 • Tay-Sachs disease. “Zebra bodies” comprised of concentric membrane-bound lysosomal inclusions (electron microscopy, 20,000×). |
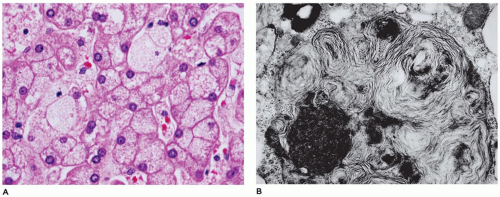 FIGURE 15-24 • Niemann-Pick disease, type C. A: Hepatocytes and Kupffer cells with swollen, granular to foamy vacuolated cytoplasm (H&E, 400×). B: Large pleomorphic membrane-bound lysosomal inclusions with concentric to parallel lamellae (electron microscopy, 15,000×). |
levels in all neonatal cholestasis), normal or only minimally elevated GGT levels, and absence of pruritus. Diagnosis is made by measurement of serum and urinary bile acid levels and detection of specific enzyme defects and/or genetic defects.
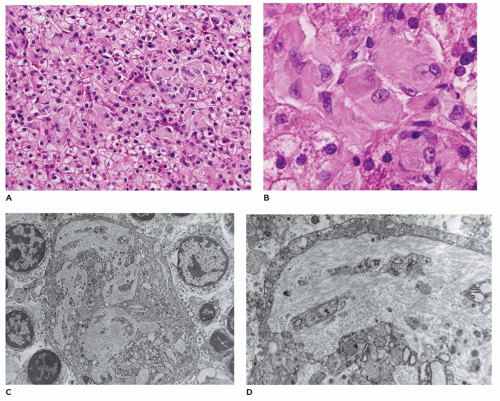 FIGURE 15-25 • Gaucher disease. A, B: Markedly enlarged Kupffer cells with cytoplasm with a striated, wrinkled tissue paper appearance (H&E, A: 100×, B: 800×). C, D: Kupffer cells with cytoplasmic tubular inclusions with a circular profile on cross-section (electron microscopy, C: 6000×, D: 24,000×). |
deficiency (congenital bile acid synthesis defect type 2). This is caused by mutation in the AKR1D1 gene (7q32-33). Congenital bile acid synthesis defect type 4 is caused by mutation in the alpha-methylacyl-CoA racemase (AMACR) gene located at 5p13.2-q11.1. Neonatal hepatitis with bile duct proliferation is associated with a bile synthesis defect in oxysterol 7-alpha-hydroxylase (CYP7B gene at 6p21.1-p11.2). Cholesterol is converted into one of several oxysterols prior to being 7-alpha-hydroxylated by an oxysterol 7-alpha-hydroxylase. Lack of this enzyme leads to neonatal hepatitis, with the potential for progressive liver disease, that can lead to infantile death. A deficiency in 25-hyroxylase (gene at 10q23) results in a bile acid synthesis defect. This is due to the role of this enzyme in expression of genes involved in cholesterol and lipid metabolism. Liver fibrosis is somewhat variable, with a more prolonged course of fibrosis in affected neonates and children. Early detection of bile acid synthetic defects can prevent progressive liver disease due to the good response to oral bile acid replacement therapy in most defects.
3 months of life (112). The stored material may be demonstrable by IHC, even in the absence of appreciable globules. Ultrastructurally, the stored material appears as flocculent, moderately electron-dense material within dilated cisternae of rough endoplasmic reticulum.
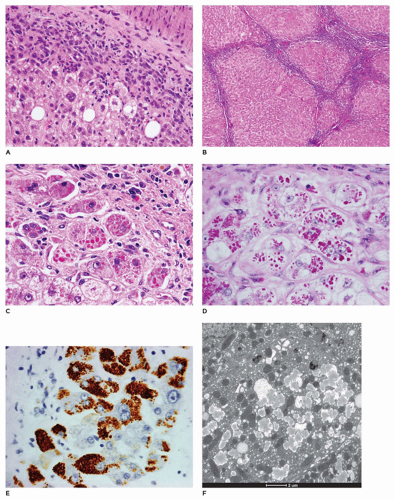 FIGURE 15-26 • A: Alpha-1 antitrypsin deficiency. Zone 1 hepatocytes with reactive changes and portal areas with chronic inflammation and mild bile duct proliferation (A: H&E, 200×). B: Cirrhosis in late stage of disease detection (B: H&E, 40×). C, D: Zone 1 hepatocytes with PAS-positive (C: 400×) cytoplasmic globules that are diastase resistant (D: 400×). E: Immunostaining for alpha-1 antitrypsin reacts with the cytoplasmic globules (E: 400×). F: Granular, flocculent material distends cisternae of the rough endoplasmic reticulum (F: electron microscopy, 10,000×). |
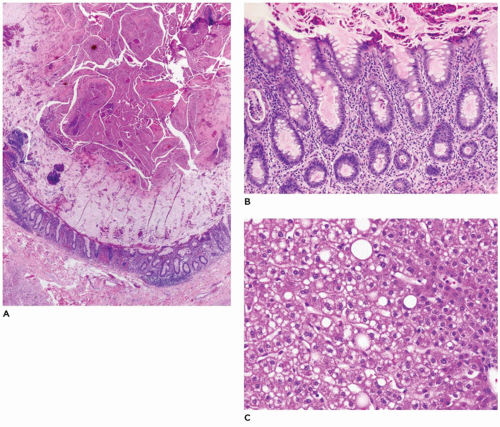 FIGURE 15-27 • Cystic fibrosis. A: Appendix with dilated lumen and dense eosinophilic mucin in the lumen (H&E, 20×). B: Appendiceal glands with inspissated densely eosinophilic mucin (H&E, 200×). C: Hepatocytes with diffuse microsteatosis and focal macrosteatosis (H&E, 200×). |
biliary cirrhosis). The progression from neonatal cholestasis to focal biliary cirrhosis is not clear.
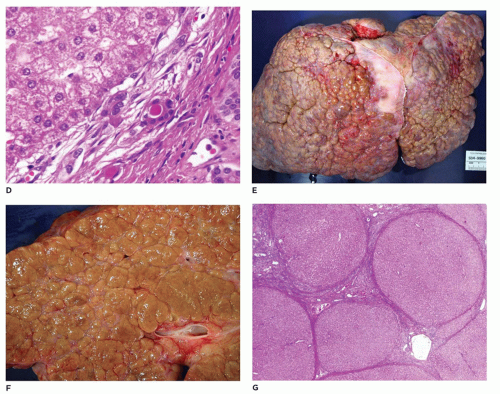 FIGURE 15-27 • (continued) D: Bile ducts in fibrotic portal region with luminal inspissated densely eosinophilic secretions (H&E, 400×). E-G: Explanted liver with macronodular and micronodular cirrhosis (H&E, G 40×). |
early, this is a form of preventable cancer. At least five iron overload disorders labeled hemochromatosis have been identified on the basis of clinical, biochemical, and genetic characteristics (6,119). Classic hemochromatosis (HFE), an autosomal recessive disorder, is most often caused by a mutation in a gene designated HFE on chromosome 6p21.3. It has also been found to be caused by a mutation in the gene encoding hemojuvelin (HJV), which maps to 1q21. Juvenile hemochromatosis or hemochromatosis type 2 (HFE2) is also autosomal recessive. One form, designated HFE2A, is caused by a mutation in the HJV gene (1q21). A second form, designated HFE2B, is caused by a mutation in the gene encoding hepcidin antimicrobial peptide, which maps to 19q13. Hemochromatosis type 3, also an autosomal recessive disorder, is caused by mutation in the gene encoding transferrin receptor 2 (TFR2), which maps to 7q22. Hemochromatosis type 4, an autosomal dominant disorder, is caused by a mutation in the SLC40A1 gene, which encodes ferroprotein and maps to 2q32. Most affected children and adolescents are asymptomatic with periportal iron accumulations extending toward the central lobule during adolescents. With juvenile hemochromatosis, organ failure with severe iron overload presents before age 30. Both the inherited (hemochromatosis) and secondary (transfusion) forms of iron storage differ from hemosiderosis in that the iron, in addition to being deposited in mononuclear phagocytic cells, is also deposited in the parenchymal cells. Iron deposition in biliary epithelial cells is seen more often in inherited iron storage disease.
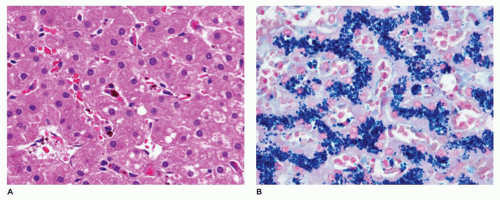 FIGURE 15-28 • Secondary hemosiderosis due to chronic transfusion therapy. A: Occasional Kupffer cells with iron pigment in their cytoplasm (H&E, 400×). B: Abundant iron storage in Kupffer cells revealed with Prussian blue histochemical stain for iron (400×). |
as bile duct epithelial cells. With other organ systems, the iron deposits tend to be within the reticuloendothelial system, with sparing of the parenchymal cells. Minor salivary glands in the oral mucosa show iron deposition in AGH and may be biopsied for diagnosis in suspected cases while awaiting genetic testing results for hemochromatosis.
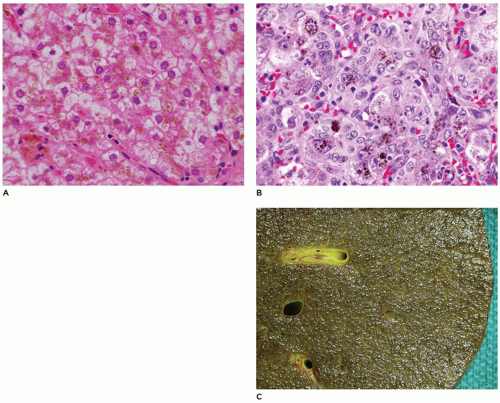 FIGURE 15-29 • Alloimmune gestational hepatitis. A, B: Hepatocytes and bile duct epithelium with readily identified iron pigment accumulation in cytoplasm, note giant cell hepatocytes (H&E, 400×). C: Explanted liver with micronodular cirrhosis and green and brown pigmentation from bile and iron accumulation, respectively. |
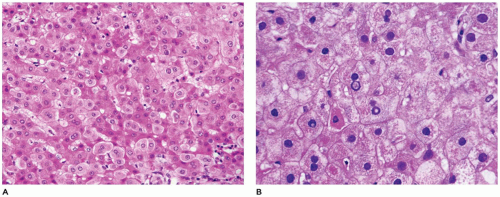 FIGURE 15-30 • Wilson disease. A: Hepatocytes with variable cytoplasmic swelling and decreased cytoplasmic eosinophilia (H&E, 200×). B: Infrequent hepatocytes with glycogenated nuclei, apoptotic (acidophil) bodies, and fine granular cytoplasm with a certain degree of cytoplasmic swelling (H&E, 400×). |
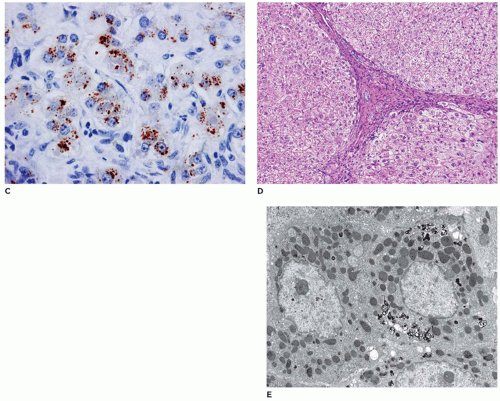 FIGURE 15-30 • (continued) C: Cytoplasmic copper detection in periportal hepatocytes (Rhodamine stain, 400×). D: Wilson disease with cirrhosis of the liver (H&E, 100×). E: Variably sized and relatively pleomorphic mitochondria and dense lysosomal deposits in Wilson disease (electron microscopy, 3000×). |
argininosuccinic acid synthetase (9q34.1), argininosuccinic aciduria due to deficiency of argininosuccinase lyase (7cenq11.2), argininemia associated with arginase deficiency (6q23), and deficiency of N-acetyl-glutamate synthetase (17q21.31) (130,131). With the exception of ornithine transcarbamylase deficiency, which is inherited as an X-linked dominant condition, the other conditions have an autosomal recessive pattern of inheritance. Prenatal diagnosis is possible. Liver biopsy in urea cycle defects may be normal or may have only mild nonspecific changes including steatosis, cholestasis, individual cell necrosis, and early fibrosis (132) (Figure 15-31). Liver transplantation may be necessary depending upon the specific urea cycle defect disorder (133). The liver being normal during transplant, these explanted livers can be used as donor organs for other patients with liver disease (domino transplant).
 FIGURE 15-31 • Urea cycle disorder—ornithine transcarbamylase deficiency. A, B: Explanted liver with no gross abnormalities in ornithine transcarbamylase deficiency. C: Portal triad and zone 1 and 2 hepatocytes with no histopathologic abnormalities (H&E, 200×). |
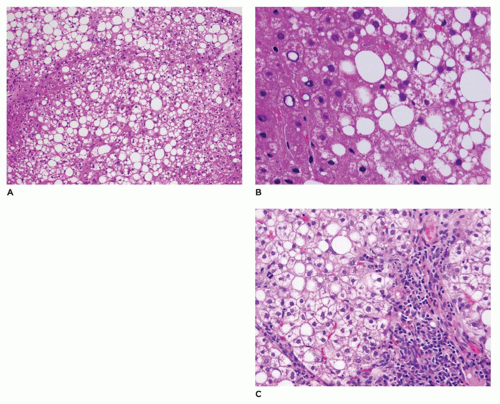 FIGURE 15-32 • Nonalcoholic fatty liver disease. A: Hepatocytes with macrovesicular and microvesicular steatosis in an azonal pattern (H&E, 100×). B: Macrosteatosis and occasional hepatocytes with glycogenated nuclei (H&E, 400×). C: Portal expansion by chronic inflammatory cells with extension into zone 1 (H&E, 200×). |
important role in the progression of liver disease with a portal-based pattern leading to liver fibrosis, especially in males. This may explain the prevalence of this disease in the pediatric age group and hence the need to identify and reverse childhood obesity to prevent progressive liver disease.
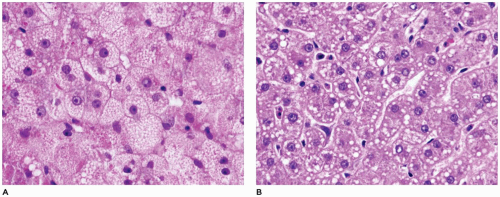 FIGURE 15-33 • Fatty acid oxidation defect—carnitine deficiency. A-C: Variable lipid deposition from fine cytoplasmic vacuolization (A) to microsteatosis (B) to macrosteatosis (C) within hepatocytes (H&E, A: 400×, B: 200×, C: 400×). D: Nonmembrane-bound lipid droplets within the hepatocyte cytoplasm (electron microscopy, 4000×). |
Between clinical episodes, the liver may appear normal. It is important to keep this group of metabolic disorders in mind when a child dies rather abruptly during a seemingly innocuous febrile illness. Tissue and fluids should be obtained at the time of autopsy and be appropriately preserved for possible biochemical and genetic analysis.
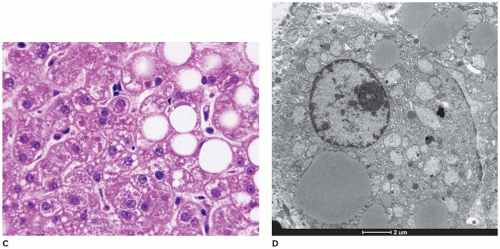 FIGURE 15-33 • (continued) |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


