I. NORMAL ANATOMY. The largest solid organ of the body, the mass of the adult liver is 1200 to 1600 g. The right, left, and caudate lobes are subdivided into segments on the basis of inflow blood supply (e-Fig. 15.1).* The liver has dual inflow supply, with approximately two-thirds from the low pressure, low O2 portal vein, and one-third from the systemic pressure, high O2 hepatic artery. Pressure equalization occurs in the sinusoids, along with the nutrient and O2 gradient from portal tracts to terminal hepatic venules. The venous return is via the left, right, and middle hepatic veins which join to form the inferior vena cava as it enters the heart at the right atrium. Bile duct blood supply is entirely from the hepatic artery plexuses.
Microscopically, the hepatic cords are lined by reticulin fibers and separated by sinusoids. The parenchyma is subdivided into acinar units of Rappaport which reflect an oxygen/nutrient gradient from most (zone 1) to least (zone 3), respectively; zone 2 is an ill-defined area in between. The anatomy of Rappaport’s units underlies many pathologic processes. The lobule, often used interchangeably with acinus, is a term based on the concept of the hexagon in which hepatic cords radiate from the central vein toward the portal tracts. Each portal tract is a fibrous matrix that contains a branch of the hepatic artery, a portal vein, a bile duct, and a poorly visualized lymphatic (e-Fig. 15.2). Larger portal tracts contain autonomic nerve fibers. Inflammatory cells are typically lacking, or are few in number. The parenchyma is separated from the portal tract at the limiting plate.
II. GROSS EXAMINATION AND SPECIMEN HANDLING
A. Needle core biopsy. Specimen handling depends on the reason for liver biopsy. After measuring and description of the number of cores, liver biopsies are wrapped in lens paper and fixed overnight; use of sponge pads is strongly discouraged because of the artifacts created during sectioning. Protocol “special” stains and six additional unstained sections are recommended for medical liver biopsies at initial preparation. Stains include three hematoxylin and eosin (H&E), a stain for collagen (trichrome or picrosirius red), reticulin, periodic acid-Schiff after diastase (PAS-d), and modified Perls’ for iron. Additional stains that must be available include copper or copper binding protein (rhodanine, orcein or Victoria blue; orcein is also useful to differentiate passive septa of collapse from active elastic fiber deposition in fibrosis); and Verhoeff van Gieson (VVG) for vessel wall architecture (Semin Diagn Pathol. 2006;23:190).
1. Tumor. Processing of biopsies for diagnosis of tumors includes three levels for H&E and six unstained for possible additional immunohistochemistry (IHC).
2. Immunocompromised patients. Biopsies from immunocompromised patients (typically solid organ or bone marrow transplant patients) may or may not require “rush” processing; clear communication with the submitting clinicians is required in these cases and fixation, grossing, and processing are tailored to the clinical needs.
3. Medical liver biopsy. The reason(s) for the liver biopsy, that is, diagnosis or confirmation; grading and staging of hepatitis; or other possible medical questions should be clearly understood prior to sign out.
4. Frozen section. Indications for frozen sections of liver core or wedge biopsies include donor liver evaluations for quantity of steatosis and/or portal inflammation; acute fatty liver of pregnancy (AFLP) for microvesicular steatosis detection by oil red O stain (from sections of liver biopsies at any stage of processing prior to xylene clearing, cut onto charged slides). Evaluation of intraoperatively encountered lesions is done from fresh tissue submitted on saline-moisturized gauze; cores are best sectioned with as little handling as possible, sectioned at 90 degrees to the long axis. Wedge biopsies may require breadloafing before sectioning. Frozen artifact creates spaces that may be challenging to distinguish from fat; thus conservative estimates of the degree of steatosis are recommended from frozen sections. If electron microscopic examination is expected for a potential metabolic disease, additional tissue should be fixed in 3% buffered glutaraldehyde.
5. Miscellaneous. Iron and copper tissue quantitation can be performed in reference laboratories directly from tissue in the paraffin block.
B. Wedge biopsy or excision. Wedge biopsy is not typically recommended for the evaluation of diffuse liver parenchymal diseases, as the subcapsular parenchyma contains fibrous extensions for 3 to 5 mm that may mimic fibrosis (e-Fig. 15.3). The subcapsular regions tend to show parenchymal collapse; elastosis may occur in this location as well as a result of chronic ischemia. Wedge excisions for super-ficial, circumscribed lesions are managed similarly to resections, as described below.
C. Segmentectomy, lobectomy, or partial hepatectomy are performed for large lesions that are not amenable to wedge excision. The surgery may or may not follow anatomic boundaries, and thus before sectioning, it is important to understand the procedure that was done; review of the imaging studies and reports is invaluable. After the type of surgery, mass, and dimensions of the specimen are recorded, the resection margin is inked, the appearance of the capsule noted, and the specimen is sliced in the axial plane at about 0.5 cm increments. Gross examination of the lesion(s) and nonlesional liver parenchyma should include color(s) (nutmeg; tan; bile-stained; hemorrhagic; yellow), viability (necrotic, nonnecrotic), texture (firm; hard; soft; spongy), and presence of nodularity. If a tumor has been preoperatively embolized via transarterial chemo-embolization (TACE), the percentage of tumor necrosis grossly should be assessed; however, only after complete submission and evaluation microscopically can it be adequately reported. Tumor sections (at least three) should demonstrate relationship of lesions to liver parenchyma, grossly visible vessels or ducts, margins (if close), and any variable areas within the tumor. Sections of nonneoplastic liver and inked resection margin(s) are submitted to evaluate underlying liver disease, vascular alterations, and margin status. One nontumor section of normal liver should be submitted and evaluated by routine special stains.
D. Total hepatectomy (explant) – performed for end-stage chronic liver disease, fulminant hepatic failure, or metabolic disorders – is followed by orthotopic liver transplantation. Radiology reports must be consulted before processing the specimen to ensure that radiographically detected lesions are sampled. The total weight, dimensions of each lobe, and capsule appearance are recorded. The hilum is completely removed, breadloafed, and submitted in toto proximally to distally without dissection. If a TIPSS stent has been placed, no attempt should be made to remove it as the spring-opened wires are not protected; rather, the hilar tissue should be dissected away. The liver is then placed on the cutting board facing up, and sectioned axially cephalad-caudad in about 0.5 cm increments. Each slice is carefully examined fresh and after fixation (especially in cirrhotic livers) for bulging, large, or discolored nodules (which are all gross features of dysplastic nodules [DNs], or small HCC) that require documentation
which includes location, number, size, color, and relation to the capsule or hilum. In their absence, two random sections from both the left and the right lobes are submitted. Only one section is necessary for routine special stains. The native and donor gallbladders are submitted as for routine cholecystectomies.
III. DIAGNOSTIC FEATURES OF COMMON NONNEOPLASTIC CONDITIONS. In the approach to liver biopsy, knowledge of the clinical information is essential. The adequacy of the biopsy should be assessed, which must be judged on the basis of the nature of the question(s) being asked. Grading and staging chronic hepatitis ideally involve a 1.5 cm core, and up to 11 portal tracts; <5 portal tracts is not optimal (Semin Diagn Pathol. 2006;23:132) (see Figs. 15.1 and 15.2).
Grading and staging schema were initially developed for comparisons of treatment for autoimmune and “nonAnonB hepatitis” trials, but they quickly transitioned to apply to chronic hepatitis (Hepatology. 2000;31:241). All systems share the assessment of portal and lobular necroinflammation for grade and fibrosis for stage (e-Figs. 15.1 and 15.2). One published system (Am J Surg Pathol. 1995;19:1409) is simple to apply and communicate clinically; this system can be applied for any form of chronic hepatitis, but is not meant for cholestatic or vascular diseases, or alcoholic or nonalcoholic fatty liver diseases (NAFLD).
Patterns of collagen deposition and architectural remodeling are frequently suggestive of the precedent injury: hepatitic, biliary, vascular, alcoholic, etc. Viral hepatitis and alcoholic hepatitis differ as the former is portal-based, and the latter is centered in zone 3, is perisinusoidal initially, and results in nodules the size of the acinus (e.g., micronodular cirrhosis). The portal-portal fibrosis of the chronic biliary diseases often results in cirrhotic remodeling with maintenance of the terminal hepatic venule in its central location; a “jig-saw” pattern is therefore suggestive of biliary disease.
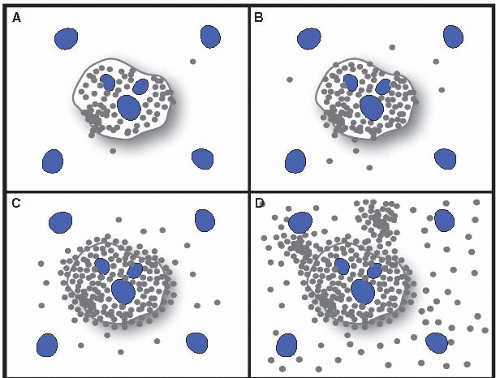
Figure 15.1 Ludwig and Batts Grading of chronic hepatitis. A: Stage 1. B: Stage 2. C: Stage 3. D: Stage 4. From: Am J Surg Pathol. 1995;19:1409. With permission.
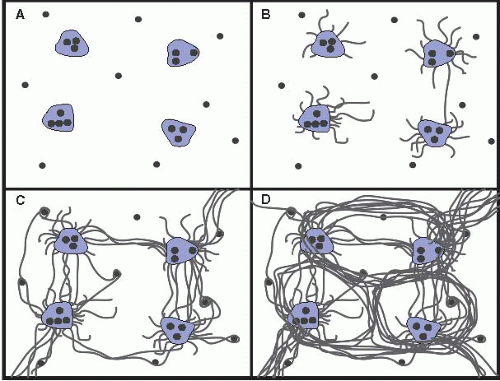
Figure 15.2 Ludwig and Batts Staging of chronic hepatitis. A: Grade 1. B: Grade 2. C: Grade 3. D: Grade 4. From: Am J Surg Pathol. 1995;19:1409. With permission.
A. Infectious liver diseases
1. Viral hepatitis refers to one of the hepatotropic viruses: HAV, HBC, HCV, HDV (with superinfection or coinfection with HBV), and HEV.
a. Acute viral hepatitis is commonly diagnosed clinically by serologic and clinical tests. Pathologists, therefore, have relatively little experience with this form of liver disease. Histopathologically, acute viral hepatitis is characterized by simultaneous hepatocyte injury and regeneration, chronic inflammation, and no fibrosis. The changes include swollen hepatocytes (hydropic degeneration), apoptotic (acidophil) bodies, lobular spotty necrosis, lobular greater than portal inflammation, sinusoidal cell reaction (Kupffer cell and endothelial cell hypertrophy), and bi- and multinucleated hepatocytes. Collectively, these findings result in “lobular unrest” and “disarray” (e-Fig. 15.4). The inflammatory infiltrates consist predominantly of mononuclear cells, with occasional plasma cells and eosinophils. In severe cases, confluent perivenular, bridging (zone 3 to zone 3), submassive (panacinar) or massive (multiacinar) hepatic necrosis may occur (e-Fig. 15.5). The ductular reaction, a form of regenerative response, may be accompanied by mononuclear or polymorphonuclear cells (e-Fig. 15.6). Trichrome and reticulin stains may be confusing as the reticulin collapse may be extensive and the collapsed sinusoids may seemingly react with the stains for collagen. In these cases, the orcein stain for elastic fibers is helpful, as elastic fibers are only found in fibrosis (e-Fig. 15.7). Zone 3 canalicular cholestasis may be present; if prominent, a diagnosis of acute cholestatic hepatitis is given (e-Fig. 15.8). Careful evaluation of portal tracts will exclude biliary obstruction (see later).
Fulminant hepatic failure or complete resolution may occur in any form of acute viral hepatitis. HAV may have zone 1 confluent or bridging necrosis and numerous plasma cells or may resemble cholestatic obstructive hepatitis. HBV, HCV, and HDV may evolve to chronic hepatitis. It is important to note that the histopathologic differential diagnoses for acute viral hepatitis include autoimmune hepatitis (AIH), Wilson’s disease, AFLP, drug-induced liver injury (DILI), and rarely, ischemic hepatitis. Cryptogenic acute hepatitis, for which no clinical cause is found, is reported as such (e-Fig. 15.9).
b. Chronic viral hepatitis, defined by persistently elevated abnormal liver tests for more than 6 months, is caused by HBV, HCV, or HDV coinfection with HBV, and is histologically characterized by mononuclear cell infiltrates rich in T lymphocytes, greater in the portal tracts than in the lobules. Plasma cells, macrophages, and eosinophils are also present, but in smaller number. The portal inflammation is accompanied by varying degrees of interface activity (previously referred to as piecemeal necrosis) (e-Fig. 15.10), lobular activity with acidophil bodies (e-Fig. 15.11) or spotty necrosis, and portal-based fibrosis (e-Fig. 15.12).
Some features are characteristic of the specific types of chronic viral hepatitis. Chronic HBV is recognized by the presence of ground glass intracytoplasmic inclusions; this is HB S Ag in expanded smooth endoplasmic reticulum (e-Fig. 15.13). HB S Ag IHC has three patterns: membranous, cytoplasmic, and inclusion (e-Fig. 15.14); intranuclear inclusions require HB C Ag IHC for identification (e-Fig. 15.15). In HBV-and HDV-coinfected cases, the necroinflammation tends to be more severe, and delta antigen can be demonstrated in nuclei by immunostaining.
The portal lymphoid aggregates of HCV are not pathognomonic because they may occur in HBV and AIH, but they are common and suggestive (e-Fig. 15.16). Steatosis is common: if in zone 1, it is most likely due to HCV; and if in zone 3, it is most likely due to host factors (metabolic or alcoholic). Acidophil bodies, bile duct injury, and sinusoidal lymphocytosis may be seen in HCV; HCV genotype 3 often has marked macrovesicular steatosis. Crystalline material is often present in substances that are abused by intravenous injection, and may be evident as polarizable material within portal macrophages.
c. Epstein-Barr virus hepatitis is characterized by a sinusoidal infiltrate of atypical lymphocytes in a “beads on a string” pattern (e-Fig. 15.17). The diagnosis can be confirmed by in situ hybridization for viral RNA and serologic tests.
d. Cytomegalovirus hepatitis in immunocompromised patients commonly induces microabscesses surrounding infected cells with characteristic intranuclear and intracytoplasmic viral inclusions (e-Fig. 15.18). Thus, microabscesses should prompt immunostaining if inclusions are not evident. CMV infection in immunocompetent patients may show microgranulomas; in this setting, viral inclusions are not usually present. Multinucleation of hepatocytes can occur in neonates.
e. Adenovirus hepatitis, uncommon in immunocompetent hosts and in adults, causes nonzonal foci of coagulative necrosis with a minimal inflammatory response. Nuclei of infected hepatocytes are hyperchromatic and smudgy, with chromatin margination (e-Fig. 15.19). Confirmation is by immunohistochemical staining.
f. Herpesvirus hepatitis includes pyknotic debris and nonzonal “punched out” necrosis; nuclear ground glass (Cowdry A) viral inclusions can be found in syncytial nuclei at the periphery of necrosis, or in other cells within the liver (e-Fig. 15.20). IHC is confirmatory. HSV is not
more common in pregnancy. Rapid diagnosis and treatment may be lifesaving.
2. Bacterial infections involve the liver in various ways: space-occupying abscess, toxic cholangitis (e.g., toxic shock syndrome), granulomatous inflammation (Mycobacterium spp.), and peliosis (bacillary angiomatosis in AIDS). Sepsis may result in microabscesses, zone 3 canalicular cholestasis, and/or ductular cholestasis (e.g., cholangitis lenta) (e-Fig. 15.21).
3. Fungal infections are due to systemic infections such as candidiasis, aspergillosis, and histoplasmosis. The biopsy may show organisms or features of sepsis (see above), or DILI.
4. Parasitic infections, including hydatid cyst, amebic abscess, and schistosomiasis, each with characteristic histopathologic features, require a high index of clinical and pathologic suspicion for diagnosis.
B. Metabolic and toxic liver diseases
1. Alcoholic liver disease (ALD) encompasses a spectrum of fatty liver, alcoholic hepatitis and steatohepatitis, alcoholic foamy degeneration, and alcoholic cirrhosis. Steatosis, steatohepatitis, and cirrhosis may be indistinguishable from NAFLD. Steatosis, reversible with abstinence, is predominantly macrovesicular and initially involves zone 3. The hallmarks of alcoholic hepatitis are hepatocytes with Mallory-Denk bodies (MDBs) and satellitosis (neutrophilic infiltration around hepatocytes containing MDB), commonly embedded in dense pericellular fibrosis (e-Fig. 15.22). The pattern of fibrosis in alcoholic hepatitis/steatohepatitis is characteristic; it begins in zone 3 as perisinusoidal/pericellular collagen deposition and eventually there is dense “chicken-wire” fibrosis that may involve the entire acinus (e-Fig. 15.23). Canalicular cholestasis may be seen. Perivenular fibrosis is common. Obliteration of the terminal hepatic venule occurs in severe alcoholic hepatitis and results in sclerosing hyaline necrosis, a poor prognostic feature (e-Fig. 15.24). Alcoholic cirrhosis is typically micronodular. Obliterated outflow veins may be found, as may copper in periseptal hepatocytes. Steatosis is variably present in ALD.
2. NAFLD may resemble some of the histologic findings of milder forms of ALD, and occurs in patients who are not heavy drinkers but who have features of metabolic syndrome (obesity, hypertension, abnormal glucose tolerance, and dyslipidemia) (Clin Liv Dis. 2010;14:591). NAFLD may also occur with a variety of medications. Unlike alcoholic hepatitis, however, a diagnosis of nonalcoholic steatohepatitis (NASH) requires the presence of steatosis; NASH does not have cholestasis and has less prominent MDB. Central sclerosing hyaline necrosis has not been reported. Minimum features of adult NASH are zone 3 macrosteatosis and hepatocyte ballooning, and lobular inflammation. Ballooning may be confirmed with K8/18 immunostaining (e-Fig. 15.25). Portal inflammation may be present in all forms of NAFLD, but if disproportionate, should raise concern of a second process such as HCV. Pediatric NAFLD may show greater steatosis, less zone-3 zonality, and more portal inflammation and fibrosis than adult NASH. The pattern of fibrosis in NASH resembles that of ALD, in that it is zone 3 perisinusoidal initially. Advanced fibrosis includes periportal fibrosis, bridging, and cirrhosis. As with ALD, steatosis may be absent in cirrhosis resultant from NASH; “burnt-out” NASH is considered the most likely cause of “cryptogenic” cirrhosis, however, other considerations include AIH and alcoholic cirrhosis (e-Fig. 15.26).
Grading and Staging NAFLD (Table 15.1). The NAFLD Activity Score (NAS) (Hepatology. 2005;41:1313), validated and developed for therapeutic trials from the original Brunt proposal for NASH, is the unweighted sum of scores for steatosis, lobular inflammation, and ballooning. The numeric NAS
and the diagnosis are separately reported, as they reflect different properties. The fibrosis score is based on trichrome stain.
TABLE 15.1 Grading and Staging of Nonalcoholic Fatty Liver Disease (NAFLD)
Component scores for grading
Steatosis
Lobular inflammation
Hepatocellular ballooning
0: <5%
0: None
0: None
1: 5-33%
1: <2/20× field
1: Mild, few
2: 34-66%
2: 2-4/20× field
2: Moderate-marked, many
3: >66%
3: >4/20 field
NAFLD activity score: NAS 0-8 (Steatosis + lobular inflammation ‘+ ballooning)
Fibrosis scoring: Based on Masson trichrome stain
0: None
1a: Delicate zone 3 perisinusoidal fibrosis, requires trichrome stain
1b: Dense zone 3 perisinusoidal fibrosis, visible on H&E
1c: Portal/periportal fibrosis
2: Zone 3 perisinusoidal and portal fibrosis
3: Bridging
4: Cirrhosis
Modified from Kleiner DE, Brunt EM, Van Natta M, et al. NIDDK sponsored NASH CRN scoring system. Hepatology. 2005;41:1313.
3. Glycogenic hepatopathy is an uncommon complication of diabetics due to poor glycemic control; hepatocytes are distended by excess glycogen as in other forms of glycogen storage disease. Steatosis and megamitochondria may be noted (e-Fig. 15.27). Diabetic hepatosclerosis is manifest by nonzonal dense perisinusoidal fibrosis; steatosis is not present. Alkaline phosphatase may be elevated (e-Fig. 15.28).
4. Iron overload (IO) is largely due to dysregulated hepcidin, the master iron regulator, which directly controls ferroportin, the macrophage exporter of iron. Production of hepcidin by hepatocytes is complex and at least partially under the control of Hfe, HJV, Tfr2, and HAMP. Hfe mutation (homozygous C282Y, the most common form of hereditary hemochromatosis [HH]), has midlife onset; HJV, Tfr2, and HAMP mutations result in younger and more severe onset. ALD, anemia of chronic disease, and the above types of hereditary HH suppress hepatocellular hepcidin with resultant hepatocyte IO.; the so-called secondary forms of IO from aberrant macrophage loading (ferroportin disease, hemophagocytic syndrome) result in macrophage sinusoidal lining cell (SLC) iron accumulation. Hepatocellular iron deposition initially occurs in a gradient from zone 1 hepatocytes, followed by the SLC. In ironloaded cirrhosis, iron-free foci are dysplastic nodules (DN); hepatocellular carcinoma (HCC) is also iron-free. Ferroportin disease and secondary IO are characterized by preferential panacinar SLC iron loading prior to hepatocellular accumulation.
Many systems exist for iron grading (Semin Liv Dis. 2005;25:392) (e-Fig. 15.29); however, diagnosis of HH requires mutational analysis as ineffective erythropoiesis also causes hepatocellular iron accumulation. Thus, histologic diagnosis should reflect only the amount of IO and a recommendation for further testing.
HH carries a significant risk of HCC. Porphyria cutanea tarda is associated with HH, steatosis, IO, cirrhosis, and also carries a risk of HCC. High ferritin levels are characteristic in NAFLD, and may raise a clinical concern of HH. Neonatal HH is a misnomer for a complement-mediated alloimmune maternal-fetal disorder with giant cell hepatitis that may or may not have liver IO (Hepatology. 2010;51:2061).
5. α1-Antitrypsin deficiency, the most common genetic pediatric liver disease, is characterized by accumulation of intracytoplasmic eosinophilic globules of varying sizes in zone 1 hepatocytes, easily demonstrated by PAS-D (e-Fig. 15.30). The diagnosis is confirmed by characteristic peripheral IHC positivity, and serum electrophoresis phenotyping. The hepatocyte globules signify a Z allele, or other rare alleles of M or S; whether or not heterozygosity is causative or enhances liver disease, is an ongoing debate (Semin Diagn Pathol. 2006;23:182). In children under 2 years of age, globules may not be present; thus, this disease is included in the differential diagnosis of giant cell or neonatal hepatitis (e-Fig. 15.31). Biliary atresia and Alagille syndromes are mimics, and it is important to remember that globules may also occur in benign and malignant liver neoplasms. The differential diagnosis of non-α1AT globules includes polyglucosan inclusions in polypharmacy; Lafora bodies; cyanamide therapy; HB S Ag; adaptation; and fibrinogen inclusions.
6. Wilson’s disease, which can occur as a result of over 200 mutations of biliary copper transporters and ceruloplasmin formation, results in copper accumulation in the liver, brain, cornea, and kidney, and presents between 5 and 45 years old. Hepatic histology is as protean as the clinical disease. Wilson’s disease can be queried for otherwise unexplained macro or microsteatosis, chronic hepatitis, cirrhosis, or submassive necrosis in a young adult. Periportal glycogenated nuclei and MDBs are common. Excessive copper may not be visible by staining as it largely remains intracytosolic; diagnostic testing requires quantitation from the paraffin block (>250 μg/g dry weight). Chronic cholestatic diseases also result in copper deposits in the eyes (Kayser-Fleischer rings) and periportal/periseptal hepatocytes, but the increase in copper does not reach the quantitative levels of Wilson’s disease (Semin Diagn Pathol. 2006;23:182).
7. Glycogen storage diseases, or glycogenoses, are characterized by abnormal accumulation of glycogen in hepatocytes giving rise to a pale, distended, and mosaic appearance. PAS stains and electron microscopy may help confirm the diagnosis.
8. Lysosomal storage diseases, the most common of which is Gaucher’s disease, are characterized by distended Kupffer cells. The cytoplasm of “Gaucher cells” is finely striated as with “wrinkled tissue paper.”
9. Hematologic disorders: Lymphoma, leukemia, hemophagocytic syndrome, and sickle cell disease. Lymphoma and leukemia are discussed further below. Macrophage activation syndrome (e-Fig. 15.32), characterized by Kupffer cell erythrophagocytosis, reflects systemic malignancy, viral infection, or collagen vascular disease. Serum ferritin levels are extremely elevated. Portal and parenchymal CD8+ lymphocytes are common. The sickled cells in sickle cell disease cause microthrombi, erythrophagocytosis, and increased hepatocellular iron (e-Fig. 15.33).
10. Reye’s syndrome (e-Fig. 15.34) and other mitochondriopathies are more common in children than adults. The liver shows pauci-inflammatory microvesicular steatosis. Ultrastructural examination highlights mitochondrial alterations. Similar changes occur in alcoholic foamy degeneration.
11. Total parenteral nutrition (TPN) results in steatosis or steatohepatitis in adults and cholestasis with a ductular reaction and fibrosis in children. In both, bridging fibrosis and cirrhosis may occur.
12. Amyloidosis commonly involves portal tract arteries and may be inconsequential, and can occur as the result of several disease entities (Clin Liver Dis 2004;8:915). However, hepatic involvement is considered terminal when amyloid replaces the sinusoids and results in compression and atrophy of the hepatocytes (e-Fig. 15.35). Trichrome stain shows the characteristic gray color of amyloid, and polarized Congo red shows the apple-green birefringence of amyloid.
13. Cystic fibrosis has hepatic manifestations in about 20% of patients. CF may present as a mimic of biliary atresia or neonatal hepatitis, or later as portal hypertension, due to bile duct mucus plugging and fibrosis. The histologic hallmarks are dense eosinophilic inspissated mucous in dilated ducts, cholangitis, ductular reaction, chronic inflammation, and fibrosis (e-Fig. 15.36). Focal biliary fibrosis occurs in up to 70% of adults and may warrant liver transplant.
14. Drug- and toxin-induced liver injury (DILI), commonly in the differential for unexplained liver test elevations, can be direct (predictable, intrinsic) or indirect (unpredictable, idiosyncratic). Direct toxicity involves agents known to produce liver damage in a dose-dependent manner; methotrexate, antibiotics, and chemotherapeutic agents are examples. Indirect toxicity is immunemediated and dose-independent; granulomatous and eosinophilic inflammation typifies this type of injury. Acute injury may lead to cholestatic hepatitis, bland cholestasis, interlobular duct damage, acute hepatitis, and massive necrosis. Chronic injury may assume the form of chronic hepatitis, granulomatous hepatitis, steatosis, steatohepatitis, vascular injury, fibrosis, cirrhosis, or neoplasia. Oxaliplatin injury is discussed below in sinusoidal obstruction syndrome (SOS).
C. AIH and bile duct disorders of the liver
1. Autoimmune hepatitis (AIH) can present in adolescent or postmenopausal females, and is associated with hypergammaglobulinemia (IgG) and high titers of antinuclear and antismooth muscle antibodies in adults, and antiliver-kidney microsomal type 1 antibodies in girls. The diagnosis should only be made after other metabolic diseases have been excluded, and after negative viral serologies have been demonstrated (Hepatology. 2008;48:169). In classic cases, there is a dense portal and lobular mononuclear cell infiltrate enriched in plasma cells (e-Fig. 15.37). Marked interface hepatitis, centrilobular confluent or bridging necrosis, and hepatitic rosetting are present. Advanced fibrosis may be found at presentation. Cholestasis is rare but signifies severity. AIH may rarely present as fulminant hepatic failure. Likewise, mild chronic hepatitis and/or cirrhosis may be the initial findings.
2. Primary biliary cirrhosis (PBC), a progressive cholestatic disease of middleaged women, results in the destruction of intrahepatic bile ducts. IgM and serum cholesterol are elevated. The early stage (Table 15.2) has mixed portal inflammatory infiltrates and the pathognomonic florid duct lesion consisting of granulomatous or lymphocytic infiltration of duct epithelium (e-Fig. 15.38). The granulomas in PBC are epithelioid, may be portal or lobular, and present in any stage of disease. Stains for fungal and acid-fast organisms are appropriate at the time of initial diagnosis. The disease is inhomogenous, but ductular reaction, interface hepatitis, chronic cholestasis, and biliary piecemeal necrosis develop with progression, with eventual bridging necrosis, septal fibrosis, and biliary cirrhosis. Chronic cholestasis (cholate stasis) is characterized by periportal edema, ductular reaction, MDBs, lobular foam cells, copper (e-Fig. 15.39), and cholestatic rosettes. Nodular regenerative hyperplasia (NRH)-like parenchymal features may occur in any stage of PBC and may explain the clinical findings of noncirrhotic variceal bleeding.
Autoimmune cholangiopathy and anti-mitochondrial antibody (AMA) negative PBC are synonymous terms for seronegative PBC that is otherwise clinically and histologically identical to PBC (and is distinct from overlap syndrome as discussed below). ANA testing is often positive in this setting.
3. Primary sclerosing cholangitis (PSC), a progressive fibro-obliterative disorder primarily of young men, is of unknown etiology. PSC affects the extra- and intrahepatic biliary tree leading to biliary strictures and ectasias, and cirrhosis. PSC is strongly associated with ulcerative colitis. The definitive diagnosis
of PSC rests on imaging by MRCP or ERCP; liver biopsy may be suggestive (features of chronic cholestasis) or confirmatory (periductal fibrosis), but is not the diagnostic test (see later). Fibrous cholangitis (the so-called onionskin fibrosis) involving large and/or small bile ducts, duct damage, ductular reaction, and chronic cholestasis are typical findings (e-Fig. 15.40). PSC inhomogenously progresses to cirrhosis and is staged according to portal changes (Table 15.2). Cholangiocarcinoma occurs in up to 10% of cases of PSC. Currently, carefully selected and protocolized patients with PSC and cholangiocarcinoma undergo neoadjuvant therapy and months of surveillance prior to transplant.
TABLE 15.2 Grading and Staging
Grading and staging of primary biliary cirrhosisa
Stage
Scheuer
Ludwig
1
Florid duct lesion
Portal stage
Bile duct damage
Portal inflammation
Portal inflammation
2
Ductular proliferation (reaction)
Periportal stage
Expanded portal tracts
Periportal inflammation ± piecemeal necrosis
Proliferated ductules
Piecemeal necrosis
3
Scarring
Septal stage
Fibrosis
Fibrous septa
Loss of ducts
Bridging necrosis
4
Nodular cirrhosis
Cirrhosis
Grading and staging primary sclerosing cholangitisb
Stage 1
Portal stage
Portal inflammation or bile duct abnormalities
Stage 2
Periportal stage
Periportal fibrosis or enlargement of portal tract
Stage 3
Septal stage
Fibrous septal or bridging necrosis
Stage 4
Cirrhotic stage
Cirrhosis
a Modified from Lee R., ed. Diagnostic Liver Pathology. 1994, p. 117 .
b Modified from Lee R., ed. Diagnostic Liver Pathology. 1994, p. 127 .
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


The Liver
The Liver
Ta-Chiang Liu
Elizabeth M. Brunt
