The blood urea nitrogen (BUN) is a better indicator of uremic symptoms than the serum creatinine which is a better indicator of glomerular filtration rate (GFR).
 The BUN reflects protein intake, so a patient who has not been eating may have a BUN that is less reflective of renal function.
The BUN reflects protein intake, so a patient who has not been eating may have a BUN that is less reflective of renal function.
 In the presence of volume depletion BUN is affected more than serum creatinine since decreased renal blood flow is associated with back diffusion of urea (urea clearance is affected more than creatinine clearance).
In the presence of volume depletion BUN is affected more than serum creatinine since decreased renal blood flow is associated with back diffusion of urea (urea clearance is affected more than creatinine clearance).
The ratio of BUN to creatinine, normally about 10 to 1, is increased in prerenal azotemia.
 Urinary tract obstruction, which can be easily ruled out by imaging techniques, is also associated with an elevated BUN/creatinine ratio.
Urinary tract obstruction, which can be easily ruled out by imaging techniques, is also associated with an elevated BUN/creatinine ratio.
Plasma Volume Assessment
The clinical assessment of volume status is crucial in the severely ill patient. Neck vein distention 3 cm above the clavicle at 30 degrees of elevation indicates an increased filling pressure. Edema is a critical sign. In a bed-ridden patient edema of the posterior thighs may be more reliable than pedal or pretibial fluid.
 Edema always means excess fluid; it equilibrates well and rapidly with the plasma volume.
Edema always means excess fluid; it equilibrates well and rapidly with the plasma volume.
Exceptions are intense arteriolar vasodilation without venodilation (e.g., calcium channel blockers, cytokine release in septic shock), venous insufficiency, and extremely low albumin levels. With these absent, there is no such thing as “volume depletion” in an edematous patient.
 The body assesses volume status from changes in pressure in the capacitance (low pressure) portion of the circulation (great veins and right atrium).
The body assesses volume status from changes in pressure in the capacitance (low pressure) portion of the circulation (great veins and right atrium).
The body is good at assessing changes in pressure but unable to assess changes in volume, so it uses a surrogate to estimate plasma volume based on pressure in the capacitance vessels. A decrease in plasma volume decreases venous return which decreases the pressure (stretch) in the great veins and the right atrium; this decrease in pressure stimulates the sympathetic nervous system causing venoconstriction which returns blood from the periphery to the central venous reservoir, thereby compensating for the volume deficit and maintaining cardiac output. This occurs at the expense of a diminished plasma volume. At the same time activation of the renin-angiotensin-aldosterone system (RAAS) increases renal sodium reabsorption tending to restore extracellular fluid volume.
 The entity of “stress polycythemia” is really hemoconcentration around a normal red cell mass.
The entity of “stress polycythemia” is really hemoconcentration around a normal red cell mass.
Conversely, an increase in plasma volume increases the pressure (and stretch) in the capacitance portion of the circulation, diminishes sympathetic activity, suppresses the RAAS, and initiates a diuresis, thereby diminishing the extracellular fluid volume.
HYPONATREMIA
Volume Depletion Hyponatremia
 Hyponatremia in the presence of volume depletion represents the triumph of volume over tonicity (Table 6-1).
Hyponatremia in the presence of volume depletion represents the triumph of volume over tonicity (Table 6-1).
TABLE 6.1 Hyponatremia
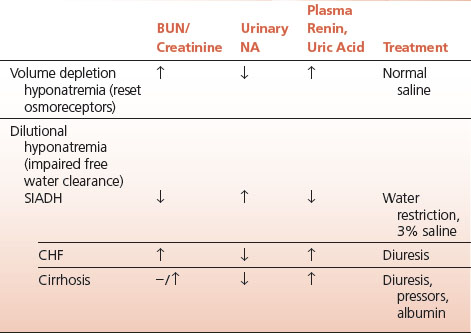
Low-serum sodium is either dilutional or secondary to diminished plasma volume. In the latter, as plasma volume diminishes neural afferent impulses from the low-pressure capacitance portion of the circulation change the normal relationship between tonicity and antidiuretic hormone (ADH) release so that ADH is released at lower plasma tonicity than normal. This represents a resetting of the central osmoreceptors. Volume is thereby preserved at the expense of tonicity.
 In volume depletion hyponatremia the urine is concentrated, the urinary sodium is low, and plasma renin and serum uric acid are high, reflecting diminished renal blood flow.
In volume depletion hyponatremia the urine is concentrated, the urinary sodium is low, and plasma renin and serum uric acid are high, reflecting diminished renal blood flow.
Volume depletion hyponatremia thus represents the wisdom of the body in defending an adequate circulatory volume at the expense of maintaining normal tonicity. Treatment thus entails volume resuscitation with normal saline that permits a water diuresis and the prompt restitution of the serum sodium.
Dilutional Hyponatremia
 The essential abnormality of dilutional hyponatremia is the inability to have a water diuresis in the presence of an increased extracellular fluid volume and a low plasma tonicity.
The essential abnormality of dilutional hyponatremia is the inability to have a water diuresis in the presence of an increased extracellular fluid volume and a low plasma tonicity.
Dilutional hyponatremia is complicated in both pathophysiology and treatment.
Syndrome of Inappropriate Secretion of ADH (SIADH)
The purest form of dilutional hypernatremia is the SIADH. The inappropriate ADH may be synthesized and secreted ectopically by tumors (lung or other), released from the posterior pituitary by poorly characterized reflex mechanisms, or result from the action of drugs which potentiate ADH release.
 A very low-serum uric acid may be a tip off to the diagnosis of SIADH.
A very low-serum uric acid may be a tip off to the diagnosis of SIADH.
 Intrathoracic lesions such as pneumonia or CNS lesions such as strokes are common causes of SIADH.
Intrathoracic lesions such as pneumonia or CNS lesions such as strokes are common causes of SIADH.
In SIADH the tonicity of urine usually exceeds that of plasma and in all cases is less than maximally dilute. The urinary sodium concentration is high, reflective of the daily salt intake.
 The natriuresis that occurs in SIADH once again represents the triumph of volume over tonicity, since the plasma volume expansion induced by water reabsorption is countered by salt excretion.
The natriuresis that occurs in SIADH once again represents the triumph of volume over tonicity, since the plasma volume expansion induced by water reabsorption is countered by salt excretion.
In the past this led to the outmoded concepts of “pulmonary” or “cerebral” salt wasting since the high urinary sodium was recognized before the physiology of the dilutional state was understood. The treatment is fluid restriction (below insensible loss) and hypertonic saline if the hyponatremia is severe or symptomatic.
 Other forms of dilutional hyponatremia that may occur with clinically obvious expansion of the extracellular fluid space include congestive heart failure (CHF) and cirrhosis.
Other forms of dilutional hyponatremia that may occur with clinically obvious expansion of the extracellular fluid space include congestive heart failure (CHF) and cirrhosis.
In these cases it is likely that a primary decrease in renal blood flow impairs the ability to clear free water. This is supported by a high plasma renin and elevated serum uric acid, both reflections of diminished renal perfusion. Treatment is aimed at restoring renal perfusion so that a water diuresis is possible.
 Thiazide-induced hyponatremia has features of both volume depletion and dilution.
Thiazide-induced hyponatremia has features of both volume depletion and dilution.
It rapidly normalizes after discontinuation of the thiazide diuretic.
 The hyponatremia of Addison’s disease reflects both hypovolemia (deficient aldosterone) and the fact that a permissive level of glucocorticoid is necessary to achieve maximal impermeability of the distal nephron to water in the absence of ADH.
The hyponatremia of Addison’s disease reflects both hypovolemia (deficient aldosterone) and the fact that a permissive level of glucocorticoid is necessary to achieve maximal impermeability of the distal nephron to water in the absence of ADH.
Thus, back diffusion of water limits the ability to clear free water. This explains the hyponatremia of secondary adrenal insufficiency which occurs in spite of unimpaired aldosterone secretion.
 Hyponatremia is common in hypopituitarism despite normal aldosterone secretion since permissive amounts of glucocorticoids and thyroid hormones are necessary for a normal water diuresis.
Hyponatremia is common in hypopituitarism despite normal aldosterone secretion since permissive amounts of glucocorticoids and thyroid hormones are necessary for a normal water diuresis.
URINARY SODIUM AND POTASSIUM
Sodium and Potassium Balance
 In the absence of disease a state of sodium and potassium balance exists: excretion reflects dietary intake. Nothing is gained by measuring these cations unless you are assessing the renal response to a physiologic perturbation such as hyponatremia or hypokalemia.
In the absence of disease a state of sodium and potassium balance exists: excretion reflects dietary intake. Nothing is gained by measuring these cations unless you are assessing the renal response to a physiologic perturbation such as hyponatremia or hypokalemia.
In volume depletion, for example, the kidney conserves sodium and the urinary sodium will be very low. In dilutional forms of hyponatremia the sodium will be high (reflective of intake).
 In patients with hypokalemia a spot urine potassium measurement is very useful: a urinary potassium level below 20 mEq/L suggests extrarenal losses; higher urinary potassium indicates renal loss, usually secondary to increased aldosterone secretion, which induces potassium and hydrogen ion excretion in exchange for sodium reabsorption.
In patients with hypokalemia a spot urine potassium measurement is very useful: a urinary potassium level below 20 mEq/L suggests extrarenal losses; higher urinary potassium indicates renal loss, usually secondary to increased aldosterone secretion, which induces potassium and hydrogen ion excretion in exchange for sodium reabsorption.
ACID–BASE DISTURBANCES
Metabolic Acidosis
 In the absence of toxic ingestion or advanced renal failure an anion gap acidosis is either lactic acidosis or ketoacidosis. If measurements for ketones in the blood are negative, lactic acid is the cause.
In the absence of toxic ingestion or advanced renal failure an anion gap acidosis is either lactic acidosis or ketoacidosis. If measurements for ketones in the blood are negative, lactic acid is the cause.
The causes of lactic acidosis are many, but poor perfusion and tissue necrosis, often in the setting of multiple organ failure, are frequent contributors. Ketoacidosis is caused by hepatic ketone overproduction, the latter a consequence of insulin deficiency at the level of both the liver and adipose tissue depots throughout the body. Circulating free fatty acids (FFAs) are the substrate for ketone production. Low levels of insulin, as found in the fasting state, encourage lipolysis in adipose tissue and the increased generation of FFAs. In the liver, the FFAs are converted into ketoacids (β-hydroxybutyrate and acetoacetate) rather than synthesized into triglycerides because of the low levels of insulin which puts the liver in a ketogenic mode.
 There is a difference between low levels of insulin and absent insulin: in fasting, insulin, although low, is present in sufficient quantities to prevent the unrestrained lipolysis and enhanced delivery of FFAs to the liver that characterizes the absence of insulin.
There is a difference between low levels of insulin and absent insulin: in fasting, insulin, although low, is present in sufficient quantities to prevent the unrestrained lipolysis and enhanced delivery of FFAs to the liver that characterizes the absence of insulin.
This is the difference between simple starvation and diabetic ketoacidosis (DKA); in the former, ketosis occurs but not ketoacidosis; in the latter, the total absence of insulin greatly enhances lipolysis which provides the substrate (FFAs) for unrestrained ketogenesis in liver with consequent ketoacidosis.
 Alcoholic ketoacidosis occurs in the setting of poor nutrient intake coupled with the very severe volume depletion that complicates serious alcohol abuse.
Alcoholic ketoacidosis occurs in the setting of poor nutrient intake coupled with the very severe volume depletion that complicates serious alcohol abuse.
In this setting insulin secretion is suppressed by the sympathetic stimulation that accompanies alcohol-induced fluid losses (vomiting and diarrhea). SNS stimulation strongly inhibits insulin secretion by an α-adrenergic mechanism exerted on the pancreatic beta cells. In addition, the metabolism of ethanol increases the concentration of acetyl CoA in the liver by blocking entrance into the Krebs cycle and by blocking the oxidation of FFAs, thus enhancing ketone body formation.
 In distinction to DKA, alcoholic ketoacidosis is characteristically mild and responds readily to fluid resuscitation.
In distinction to DKA, alcoholic ketoacidosis is characteristically mild and responds readily to fluid resuscitation.
Other states of severe volume depletion may be associated with significant ketosis and ketoacidosis as well. Hyperemesis gravidarum and acute pancreatitis are common examples.
 In the presence of advanced renal insufficiency metformin may cause lactic acidosis.
In the presence of advanced renal insufficiency metformin may cause lactic acidosis.
Lactic acidosis was much more common with phenformin, the predecessor of metformin.
 The “delta gap” or the “delta-delta” calculation identifies a pre-existing acid–base disturbance in the presence of an acute anion gap acidosis.
The “delta gap” or the “delta-delta” calculation identifies a pre-existing acid–base disturbance in the presence of an acute anion gap acidosis.
By subtracting the normal anion gap (12) from the calculated anion gap (say 20) and adding the difference (8) to the measured bicarbonate concentration (say 22) it is possible to estimate the serum bicarbonate (30) at the onset of the anion gap acidosis, thereby identifying a pre-existing metabolic alkalosis.
Acidosis in Renal Disease
 Acidosis in the presence of renal disease is not associated with an increased anion gap until the renal disease is end stage.
Acidosis in the presence of renal disease is not associated with an increased anion gap until the renal disease is end stage.
Acidosis in renal disease is a consequence of either renal tubular dysfunction or the loss of sufficient renal mass to excrete the acid burden generated on a daily basis.
 In renal tubular acidosis (RTA) the urine cannot be acidified to the normal extent (pH below 5.3) because of either failure to reclaim bicarbonate from the glomerular filtrate (proximal RTA, type 2 RTA) or a defect in the excretion of hydrogen ions (distal RTA, type 1 RTA). The anion gap is normal. Type 4 RTA is aldosterone deficiency which prevents the distal acidification of the urine.
In renal tubular acidosis (RTA) the urine cannot be acidified to the normal extent (pH below 5.3) because of either failure to reclaim bicarbonate from the glomerular filtrate (proximal RTA, type 2 RTA) or a defect in the excretion of hydrogen ions (distal RTA, type 1 RTA). The anion gap is normal. Type 4 RTA is aldosterone deficiency which prevents the distal acidification of the urine.
In the presence of severe reduction in GFR, below 25% of normal, plasma bicarbonate falls and chloride rises resulting in metabolic acidosis with a normal anion gap.
 The acidosis in advanced renal failure is caused by insufficient ammonium production to neutralize the daily acid burden.
The acidosis in advanced renal failure is caused by insufficient ammonium production to neutralize the daily acid burden.
The urinary pH is appropriately low in renal failure and the ammonium production per functioning nephron is high, but there are insufficient nephrons to produce the adequate ammonium and the plasma pH falls. In more advanced renal failure the anion gap does increase as the retention of organic acids and phosphate increases and the chloride levels fall.
Metabolic Alkalosis
 Hypokalemic alkalosis, especially when severe and poorly responsive to potassium repletion, reflects mineralocorticoid excess.
Hypokalemic alkalosis, especially when severe and poorly responsive to potassium repletion, reflects mineralocorticoid excess.
Hyperaldosteronism, either primary or secondary, is the usual but not the exclusive cause. These two forms of aldosterone excess are easily distinguished by plasma renin assay: suppressed in primary, elevated in secondary.
 Potassium chloride is the treatment of choice for metabolic alkalosis.
Potassium chloride is the treatment of choice for metabolic alkalosis.
Metabolic alkalosis usually occurs in the setting of chronic volume contraction and diminished renal perfusion, frequently a consequence of diuretic administration; the kidney, which is avidly reabsorbing sodium, must secrete hydrogen and potassium ions in exchange for sodium once chloride in the filtrate is completely reabsorbed. The result is inappropriately acidified urine in association with systemic alkalosis. The goal of treatment is to restore the ability of the kidney to sustain an alkaline diuresis by providing chloride, which permits sodium to be reabsorbed isoelectrically with the chloride. Since sodium is frequently inappropriate in the clinical setting, potassium chloride is the treatment of choice, since it also corrects the commonly associated potassium deficit.
Respiratory Alkalosis
 Respiratory alkalosis occurs acutely in the hyperventilation syndrome (psychogenic, panic attacks), salicylate intoxication, and sepsis, and chronically in hepatic encephalopathy.
Respiratory alkalosis occurs acutely in the hyperventilation syndrome (psychogenic, panic attacks), salicylate intoxication, and sepsis, and chronically in hepatic encephalopathy.
A positive Chvostek sign is a tip off to the diagnosis.
INTRINSIC RENAL DISEASE
Disease of the kidney may affect predominately the glomeruli or the interstitium. Both may result in chronic renal failure with small shrunken scarred kidneys. Statistically, glomerular diseases are more commonly associated with end stage renal failure.
Tubulointerstitial Disease
 Interstitial renal disease is usually the result of toxins or hypersensitivity reactions to drugs. Obstructive uropathy and hypertensive nephrosclerosis also cause interstitial scarring and fibrosis.
Interstitial renal disease is usually the result of toxins or hypersensitivity reactions to drugs. Obstructive uropathy and hypertensive nephrosclerosis also cause interstitial scarring and fibrosis.
Involvement initially spares the glomeruli, but eventually, scarring may involve the whole kidney and result in advanced renal failure. Tubular dysfunction with loss of renal concentrating ability may be prominent.
 In distinction to glomerular diseases, proteinuria is absent or scant, and hematuria is not prominent in interstitial renal disease.
In distinction to glomerular diseases, proteinuria is absent or scant, and hematuria is not prominent in interstitial renal disease.
Drugs commonly involved include the penicillins, cephalosporins, NSAIDs, anticonvulsants, and analgesics. In the allergic form eosinophils may be present in the urine and increased in the peripheral blood.
 In malignant hypertension proteinuria is nephrotic range and microscopic hematuria is common.
In malignant hypertension proteinuria is nephrotic range and microscopic hematuria is common.
Fibrinoid arteriolar necrosis in the renal circulation is the cause.
Glomerulonephritis
 Glomerulonephritis is the consequence of an immune attack on the kidney that results in damage and/or inflammation of the glomeruli. It may be primary or secondary, acute or chronic, focal or global; the glomerular epithelium, endothelium, or the basement membrane may be the primary target of the attack.
Glomerulonephritis is the consequence of an immune attack on the kidney that results in damage and/or inflammation of the glomeruli. It may be primary or secondary, acute or chronic, focal or global; the glomerular epithelium, endothelium, or the basement membrane may be the primary target of the attack.
The autoimmune response is composed of different mechanisms in different disease entities. These include antibodies directed against the basement membrane of the glomerulus (membranous glomerulonephritis, Goodpasture syndrome); immune complex deposition from circulating or locally derived antigen–antibody interactions (IgA nephropathy, poststreptococcal glomerulonephritis (PSGN), lupus nephritis, serum sickness, subacute bacterial endocarditis); and so-called pauci-immune glomerulonephritis in which antigen–antibody complexes cannot be demonstrated (Wegener’s, microscopic polyangiitis, ANCA-associated vasculitides).
 Fixation and activation of complement contributes importantly to the glomerular injury in immune complex glomerulonephritis.
Fixation and activation of complement contributes importantly to the glomerular injury in immune complex glomerulonephritis.
There is a corresponding decrease in circulating complement levels in renal diseases associated with immune complex deposition such as lupus nephritis and PSGN.
 The clinical manifestations of glomerulonephritis are proteinuria and/or hematuria, hypertension, and in some cases edema and renal failure.
The clinical manifestations of glomerulonephritis are proteinuria and/or hematuria, hypertension, and in some cases edema and renal failure.
The red cell cast is virtually diagnostic of glomerular injury since it is formed in the renal tubules from protein and RBCs, thereby unequivocally establishing the provenance of the hematuria at the level of the injured glomerulus. Some noninflammatory glomerular lesions produce only protein loss without hematuria.
 Renal biopsy is the usual means of identifying the various causes of glomerulonephritis, important since identification of the cause may have important therapeutic implications.
Renal biopsy is the usual means of identifying the various causes of glomerulonephritis, important since identification of the cause may have important therapeutic implications.
Secondary glomerulonephritis occurs when renal involvement is part of a generalized disease such as SLE or Wegener’s.
 PSGN is a prototypical immune complex glomerulopathy.
PSGN is a prototypical immune complex glomerulopathy.
A disease principally of children and less common today in the United States than previously, PSGN is still common worldwide and does occur in adults. The immunologic response to a group A beta-hemolytic streptococcal infection forms the immune complexes that fix complement with resultant glomerular injury.
 In distinction to acute rheumatic fever, which typically follows severe pharyngitis of any streptococcal serotype, PSGN is caused by specific nephritogenic strains, principally involving the skin.
In distinction to acute rheumatic fever, which typically follows severe pharyngitis of any streptococcal serotype, PSGN is caused by specific nephritogenic strains, principally involving the skin.
 Impetigo, or any impetiginized skin lesions with the appropriate nephritogenic strain of streptococci, can result in PSGN, indicating the importance of antibiotic treatment to eliminate the offending organism.
Impetigo, or any impetiginized skin lesions with the appropriate nephritogenic strain of streptococci, can result in PSGN, indicating the importance of antibiotic treatment to eliminate the offending organism.
The prognosis for full recovery of renal function in children is good, less so in adults.
 IGA nephropathy is the most common cause of primary glomerulonephritis in adults.
IGA nephropathy is the most common cause of primary glomerulonephritis in adults.
The immune deposits are autoantibodies to altered IgA. The glomerulonephritis is characterized by proteinuria and, frequently, gross hematuria. The glomerulonephritis may be self-limited or may progress to end stage renal disease.
 In children and young adults Henoch–Schönlein purpura is a generalized form of IgA deposition in which the skin (palpable purpura) and other organs are involved as well as the kidney.
In children and young adults Henoch–Schönlein purpura is a generalized form of IgA deposition in which the skin (palpable purpura) and other organs are involved as well as the kidney.
IgA deposition can be demonstrated in skin biopsy of the purpuric lesions.
Nephrotic Syndrome
Nephrotic syndrome refers to heavy proteinuria (in excess of 3 g/day) of any cause, and the complications pursuant to the loss of protein, of which there are many.
 The causes include diabetic nephropathy (Kimmelstiel–Wilson disease also known as diabetic nodular glomerular sclerosis), various forms of primary and secondary glomerulonephritis, amyloidosis (AL and AA, primary and secondary amyloidosis respectively), and a large number of infections, drugs, toxins, and allergens.
The causes include diabetic nephropathy (Kimmelstiel–Wilson disease also known as diabetic nodular glomerular sclerosis), various forms of primary and secondary glomerulonephritis, amyloidosis (AL and AA, primary and secondary amyloidosis respectively), and a large number of infections, drugs, toxins, and allergens.
 The clinical manifestations are edema, ascites, hyperlipidemia, and coagulopathies.
The clinical manifestations are edema, ascites, hyperlipidemia, and coagulopathies.
The pathogenesis of fluid accumulation reflects the decreased oncotic pressure of the plasma generated by the hypoalbuminemia and enhanced renal sodium reabsorption mediated at least in part by secondary aldosteronism. Protein synthesis in the liver is enhanced, including lipoprotein synthesis which, in conjunction with loss of lipoprotein lipase in the urine, results in hyperlipidemia. Loss of immunoglobulins results in increased susceptibility to infection, and loss of antithrombin III in the urine predisposes to venous thrombosis.
Treating the underlying cause, if possible, is frequently efficacious. Symptomatic treatment includes diuretics (loop plus spironolactone), statins, anticoagulation where appropriate, and aggressive surveillance for and treatment of infections.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


