The underlying hemodynamic abnormality in heart failure is an increase in ventricular end diastolic pressure.
Many descriptors have been applied to the failing heart based on the presumed underlying physiology: left heart failure, right heart failure, forward failure, congestive failure, diastolic dysfunction (heart failure with preserved ejection fraction), systolic failure (heart failure with diminished ejection fraction), to name a few. The common denominator is a rise in end diastolic pressure. This rise in pressure is transmitted backward to the atria and feeding veins resulting in pulmonary or systemic congestion with attendant dyspnea and edema.
 CHF is associated with increased sympathetic nervous system (SNS) activity. Tachycardia and enhanced cardiac contractility increase cardiac output.
CHF is associated with increased sympathetic nervous system (SNS) activity. Tachycardia and enhanced cardiac contractility increase cardiac output.
The SNS stimulation is advantageous up to a point; if overly intense the resulting tachycardia and force of contraction may become disadvantageous by increasing the myocardial need for oxygen; hence the rationale for β-blockers. Antagonizing the inotropic effect of the SNS, however, may worsen failure, necessitating care in the use of β-blockers in patients with CHF.
 A sign of CHF, hepatojugular reflux, is a manifestation of increased SNS tone.
A sign of CHF, hepatojugular reflux, is a manifestation of increased SNS tone.
When pressure is exerted on a congested liver, blood is forced into the superior vena cava; this increased volume in the capacitance veins results in a rise in venous pressure and an upward distention of the column of blood in the jugular veins. The increase in SNS activity increases the venous tone, so the jugular veins cannot accommodate the influx of blood and the height of the column rises.
 In the setting of myocardial infarction (MI) tachycardia indicates incipient or overt CHF.
In the setting of myocardial infarction (MI) tachycardia indicates incipient or overt CHF.
The rapid heart rate responds to treatment of heart failure.
 Dyspnea on exertion suggests heart failure; dyspnea at rest suggests pulmonary disease.
Dyspnea on exertion suggests heart failure; dyspnea at rest suggests pulmonary disease.
Exertion demands an increase in cardiac output. The failing heart does not have the requisite reserve to address the increased requirement and the resulting pulmonary congestion is sensed as dyspnea.
 Paroxysmal nocturnal dyspnea (PND), when classic, indicates left heart failure.
Paroxysmal nocturnal dyspnea (PND), when classic, indicates left heart failure.
 PND refers to an awakening from sleep after 2 hours with shortness of breath. The patient gets up, walks about, sometimes to an open window, and frequently spends the rest of the night in a chair.
PND refers to an awakening from sleep after 2 hours with shortness of breath. The patient gets up, walks about, sometimes to an open window, and frequently spends the rest of the night in a chair.
The pathophysiology is simple: reabsorption of edema fluid that has accumulated in the interstitium of the lower limbs into the circulation produces an endogenous volume load on the left ventricle. Orthopnea, although an important sign of CHF since the increased venous return produced by recumbency is an immediate fluid challenge that precipitates pulmonary congestion, is less specific than PND because the mechanics of breathing are easier in the upright position, and a variety of other diseases that elevate the diaphragm or effect lung function in the supine position result in orthopnea.
 The cause of bilateral pleural effusions is almost always CHF.
The cause of bilateral pleural effusions is almost always CHF.
 CHF may cause a unilateral right pleural effusion; a unilateral left-sided effusion should never be ascribed to CHF until all other potential causes are eliminated.
CHF may cause a unilateral right pleural effusion; a unilateral left-sided effusion should never be ascribed to CHF until all other potential causes are eliminated.
Right-sided pleural effusions in heart failure may reflect the fact that patients with cardiomegaly sleep right side down in order to avoid the sensation of the cardiac impulse transmitted through the pillow. This dependent position during sleep may foster fluid accumulation in the right pleural space.
 Edema in CHF has both a forward (diminished renal perfusion) and backward (increased venous pressure) component.
Edema in CHF has both a forward (diminished renal perfusion) and backward (increased venous pressure) component.
Decreased renal blood flow activates the renin–angiotensin–aldosterone system (RAAS), thereby enhancing water and salt retention.
 Dilutional hyponatremia is a frequent concomitant of CHF since the diminished renal blood flow prevents a water diuresis in the presence of hypotonicity. Angiotensin II, elevated in CHF, also stimulates central thirst receptors and increases water intake.
Dilutional hyponatremia is a frequent concomitant of CHF since the diminished renal blood flow prevents a water diuresis in the presence of hypotonicity. Angiotensin II, elevated in CHF, also stimulates central thirst receptors and increases water intake.
Antagonism of the RAAS thus plays an important role in the treatment of CHF. Improving cardiac compensation with diuresis is an effective treatment for the hyponatremia that accompanies CHF.
 The left ventricle tolerates a pressure load well but a volume load poorly; the right ventricle tolerates a volume load well but a pressure load poorly.
The left ventricle tolerates a pressure load well but a volume load poorly; the right ventricle tolerates a volume load well but a pressure load poorly.
The relative wall thickness and difference in compliance of each ventricle is the likely cause of this well-established clinical observation. Thus, the thinner and more compliant right ventricle accommodates moderate left to right shunts well for long periods of time but decompensates rapidly in the face of pulmonary hypertension. The left ventricle by contrast tolerates systemic hypertension or aortic stenosis (AS) well for long periods of time but decompensates more readily in the face of volume loads like that imposed by aortic regurgitation (AR).
 The most common cause of right heart failure is left heart failure.
The most common cause of right heart failure is left heart failure.
This truism, usually explained by the facile euphemism “pressure backup,” obscures the underlying physiology by implying that the increased left-sided pressures are directly transmitted to the pulmonary arterial system resulting in pulmonary hypertension and right-sided failure. A moment’s reflection will dispel this impossibly naive notion: if pulmonary venous pressure rose to the level of the pulmonary artery all cardiopulmonary circulation would stop (blood flows down a pressure gradient) and the patient would die in pulmonary edema. So what mediates the development of pulmonary arterial hypertension when left-sided pressures rise?
 A series of poorly characterized, neural, and possibly humoral, factors, recruited by a rise in left atrial and pulmonary venous pressures, result in pulmonary artery vasoconstriction which limits blood flow into the lungs and protects against pulmonary congestion. The price to pay is a decrease in cardiac output and the imposition of a pressure load on the right ventricle (Fig. 4-1).
A series of poorly characterized, neural, and possibly humoral, factors, recruited by a rise in left atrial and pulmonary venous pressures, result in pulmonary artery vasoconstriction which limits blood flow into the lungs and protects against pulmonary congestion. The price to pay is a decrease in cardiac output and the imposition of a pressure load on the right ventricle (Fig. 4-1).
Every compensatory mechanism exacts a price; in this case, decreasing pulmonary blood flow limits cardiac output and imposes a load on the right ventricle that presages the development of right heart failure.
First recognized in the setting of mitral stenosis (MS), these reflex changes prevent pulmonary edema, but impose a pressure burden on the right ventricle which ultimately fails, since, as noted above, the right ventricle tolerates a pressure load poorly.
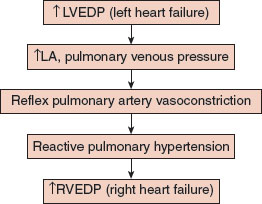
FIGURE 4.1 Left heart failure causes right heart failure. Elevation in left ventricular end diastolic pressure (LVEDP) results in increased left atrial and pulmonary venous pressures which triggers a reflex increase in pulmonary vascular resistance and a rise in pulmonary artery pressure. The pressure load on the right ventricle causes right ventricular failure.
The fact that the pulmonary hypertension is rapidly reversed by effective treatment of left heart failure demonstrates that pulmonary vasoconstriction, rather than pulmonary artery remodeling, is responsible.
 The degree of pulmonary vasoconstriction as a consequence of left heart failure varies in different patients; those with prominent vasoconstrictive responses suffer from reduced cardiac output and, eventually, right heart failure. Those with less pulmonary vasoconstriction develop pulmonary congestion and outright pulmonary edema with less manifestations of right heart failure.
The degree of pulmonary vasoconstriction as a consequence of left heart failure varies in different patients; those with prominent vasoconstrictive responses suffer from reduced cardiac output and, eventually, right heart failure. Those with less pulmonary vasoconstriction develop pulmonary congestion and outright pulmonary edema with less manifestations of right heart failure.
These series of changes occur with either diastolic (poor ventricular compliance) or systolic dysfunction (low ejection fraction) since both result in a rise in left-sided end diastolic pressures.
 Hypoxemia, an important cause of pulmonary vasoconstriction, is not the cause of the rise in pulmonary artery pressure that occurs in the setting of left heart failure.
Hypoxemia, an important cause of pulmonary vasoconstriction, is not the cause of the rise in pulmonary artery pressure that occurs in the setting of left heart failure.
The mediators of pulmonary vascular tone, in addition to oxygen, include endothelin, nitric oxide, and prostacyclin; how these are regulated in CHF remains uncertain. It is a faux pearl that hypoxia mediates the pulmonary artery vasoconstriction that accompanies left heart failure.
 Left atrial contraction, the so-called “atrial kick,” provides a given degree of ventricular stretch at a lower mean left atrial pressure than could be obtained by passive filling of the left ventricle; but for this extra stretch provided at a lower atrial pressure, pulmonary venous pressure would rise, threatening pulmonary edema.
Left atrial contraction, the so-called “atrial kick,” provides a given degree of ventricular stretch at a lower mean left atrial pressure than could be obtained by passive filling of the left ventricle; but for this extra stretch provided at a lower atrial pressure, pulmonary venous pressure would rise, threatening pulmonary edema.
The importance of atrial contraction depends upon the Frank–Starling law which relates the force of myocardial contraction to diastolic myocardial fiber length: the more stretch, the greater the force of contraction. The increase in stroke volume that accompanies an increase in venous return depends on this mechanism, which also comes into play when diastolic volume is increased in heart failure. To gain the same amount of ventricular stretch without a left atrial kick would require higher left atrial pressures.
 The significance of the atrial kick is clearly demonstrated when a patient with AS develops atrial fibrillation.
The significance of the atrial kick is clearly demonstrated when a patient with AS develops atrial fibrillation.
Decompensated CHF is the usual result. Rapid rate with diminished time for diastolic filling also contributes importantly.
 Cardiac dilatation has important adverse effects; although dilatation may be viewed as a compensatory mechanism in the face of increased end diastolic pressure, the potential benefit occurs only up to a point.
Cardiac dilatation has important adverse effects; although dilatation may be viewed as a compensatory mechanism in the face of increased end diastolic pressure, the potential benefit occurs only up to a point.
When overstretched the cardiac myocytes lose their mechanical efficiency and contract poorly. Under these circumstances the cardiac contractility declines, rather than increases, with further stretch (the so-called descending limb of Starling’s curve).
 Cardiac dilatation is detrimental for other reasons as well: increased ventricular diameter increases myocardial wall tension (Laplace’s law), and wall tension is a major determinant of myocardial oxygen consumption, thus threatening ischemia; and ventricular dilatation alters the spatial relationship of the papillary muscles to each other, so that papillary muscles no longer pull the mitral valve leaflets together resulting in mitral regurgitation (MR).
Cardiac dilatation is detrimental for other reasons as well: increased ventricular diameter increases myocardial wall tension (Laplace’s law), and wall tension is a major determinant of myocardial oxygen consumption, thus threatening ischemia; and ventricular dilatation alters the spatial relationship of the papillary muscles to each other, so that papillary muscles no longer pull the mitral valve leaflets together resulting in mitral regurgitation (MR).
CHF and Pregnancy
 The absence of CHF symptoms during pregnancy provides historical evidence of adequate cardiac reserve at that time.
The absence of CHF symptoms during pregnancy provides historical evidence of adequate cardiac reserve at that time.
Pregnancy history is important in assessing cardiac function. The 30% to 40% increase in plasma volume and cardiac output that peaks early in the third trimester provides a convenient landmark of cardiac reserve at that point in time. This is particularly important in assessing the functional significance of congenital and rheumatic valvular cardiac lesions which may become symptomatic for the first time during pregnancy.
 Peripartum cardiomyopathy is an uncommon cause of CHF occurring late in pregnancy, or more commonly, in the months immediately postpartum.
Peripartum cardiomyopathy is an uncommon cause of CHF occurring late in pregnancy, or more commonly, in the months immediately postpartum.
A dilated cardiomyopathy of unknown cause, the diagnosis of peripartum cardiomyopathy, depends on excluding all other known causes of CHF.
CARDIAC ISCHEMIA
Chest Pain
That chest pain is the hallmark of myocardial ischemia is, of course, well known. The pain is substernal, radiating to the shoulder, left arm, or jaw, frequently with sweating, and, in the presence of MI, associated with a sense of impending doom.
 Chest pain from myocardial ischemia is never fleeting; pain lasting 1 minute or less is not ischemic in origin. Pain that changes with position, or with respiration, suggests pericarditis, and not myocardial ischemia (although remember that pericarditis may complicate MI).
Chest pain from myocardial ischemia is never fleeting; pain lasting 1 minute or less is not ischemic in origin. Pain that changes with position, or with respiration, suggests pericarditis, and not myocardial ischemia (although remember that pericarditis may complicate MI).
Myocardial Ischemia and Infarction
 The determinants of myocardial oxygen consumption (MVO2) are heart rate, contractile state, and ventricular wall tension. Since tension is proportional to intraventricular pressure and ventricular radius (Laplace’s law), diastolic volume of the heart is directly related to MVO2.
The determinants of myocardial oxygen consumption (MVO2) are heart rate, contractile state, and ventricular wall tension. Since tension is proportional to intraventricular pressure and ventricular radius (Laplace’s law), diastolic volume of the heart is directly related to MVO2.
SNS stimulation increases all of these determinants, save ventricular diameter, providing the physiologic basis for the clinically established role of β-blockade in the treatment and prophylaxis of myocardial ischemia.
 Ischemia may result from diminished oxygen supply or increased demand, or both. Decreased supply occurs with the abrupt narrowing or closure of a coronary vessel while increased demand results from increases in the determinants of MVO2: tachycardia, increased BP, and/or an increase in ventricular volume.
Ischemia may result from diminished oxygen supply or increased demand, or both. Decreased supply occurs with the abrupt narrowing or closure of a coronary vessel while increased demand results from increases in the determinants of MVO2: tachycardia, increased BP, and/or an increase in ventricular volume.
MI may be transmural or subendocardial. Q waves and ST segment elevation suggest transmural infarction from an abrupt change in supply; subendocardial infarction is associated with ST segment depression in the leads reflecting the ischemic area.
 Subendocardial ischemia is most common in situations of increased demand because of the unique vulnerability of the subendocardium to alterations in perfusion.
Subendocardial ischemia is most common in situations of increased demand because of the unique vulnerability of the subendocardium to alterations in perfusion.
This vulnerability depends on the facts that the subendocardium resides at the distal end of the coronary circulation, and that it is subject to the highest wall pressure by virtue of its location next to the high-pressure ventricular cavity.
 Although usually associated with plaque rupture and acute coronary occlusion, transmural infarction may also occur with coronary vasospasm.
Although usually associated with plaque rupture and acute coronary occlusion, transmural infarction may also occur with coronary vasospasm.
Vasospasm may occur with completely normal coronaries or in association with atherosclerotic changes.
 The aim of early revascularization is to limit necrosis in the “ischemic penumbra,” the area of stunned, but still potentially viable, myocardium.
The aim of early revascularization is to limit necrosis in the “ischemic penumbra,” the area of stunned, but still potentially viable, myocardium.
PERICARDITIS
Acute Pericarditis
Acute pericarditis has many potential causes, but most often no etiology can be established resulting in the designation “idiopathic.” Most of these idiopathic cases are secondary to systemic viral infections (coxsackie or other enteroviruses). Pericarditis may also complicate intrathoracic infections with bacteria or fungi, tuberculosis being a classic example. Collagen vascular disease, post-MI, uremia, and metastatic malignancy are other possible causes.
 The pain of pericarditis is accentuated by deep breathing and cough, worse on lying supine, and ameliorated by sitting up.
The pain of pericarditis is accentuated by deep breathing and cough, worse on lying supine, and ameliorated by sitting up.
Diagnosis is secured by a friction rub and characteristic EKG changes. The rub may be brought out on examination by having the patient lean forward or kneel on all fours with the stethoscope held on the precordium. The EKG shows widespread ST elevations that often evolve into T wave inversions, PR depression, and, in the presence of a significant pericardial effusion, electrical alternans, a beat-to beat-variation in the height of the R wave as the heart swings in the fluid-filled pericardial sac.
 The feared complication of pericarditis, pericardial tamponade is suggested on physical examination (PE) and confirmed by echocardiography.
The feared complication of pericarditis, pericardial tamponade is suggested on physical examination (PE) and confirmed by echocardiography.
Pulsus paradoxus (10 mm Hg drop on inspiration), absent point of maximal impulse (PMI), distant heart sounds, and jugular venous distention (JVD) suggest the diagnosis. Cardiac echo shows fluid accumulation and collapse of the right ventricle, an indication for immediate pericardiocentesis.
 Pericarditis may be associated with MI in the immediate postinfarct period or following the infarction by weeks to months (Dressler’s syndrome). The incidence of both types of pericarditis has diminished substantially in recent years due to revascularization in the treatment of the acute event, presumably by limiting infarct size.
Pericarditis may be associated with MI in the immediate postinfarct period or following the infarction by weeks to months (Dressler’s syndrome). The incidence of both types of pericarditis has diminished substantially in recent years due to revascularization in the treatment of the acute event, presumably by limiting infarct size.
Peri-infarction pericarditis is caused by pericardial inflammation from contiguous necrotic myocardium. Almost always benign and self-limited, a developing pericardial effusion post MI is nonetheless grounds for discontinuing anticoagulation.
Dressler’s syndrome (post-MI syndrome) is an autoimmune reaction that also occurs in postsurgical patients (the postcardiotomy syndrome). Anti-inflammatory agents are useful for symptomatic control.
Chronic Constrictive Pericarditis
Chronic pericardial constriction limits the filling of all cardiac chambers and produces changes suggesting CHF.
 The presence of Kussmaul’s sign (paradoxical increase in jugular venous distention with inspiration) and ascites strongly suggest constrictive pericarditis.
The presence of Kussmaul’s sign (paradoxical increase in jugular venous distention with inspiration) and ascites strongly suggest constrictive pericarditis.
The causes of constrictive pericarditis include indolent infection (TB, fungi), malignant infiltration, collagen vascular disease, irradiation, and in many cases, unknown.
 Ascites is uncommon in CHF unless passive congestion of the liver is severe and longstanding. Right heart failure (peripheral edema, JVD) with ascites is constrictive pericarditis until proved otherwise.
Ascites is uncommon in CHF unless passive congestion of the liver is severe and longstanding. Right heart failure (peripheral edema, JVD) with ascites is constrictive pericarditis until proved otherwise.
The cause of the ascites is obscure but may reflect lymphatic obstruction at the level of the liver.
 Diagnosis of pericardial constriction may be difficult since imaging is frequently inconclusive; pericardial calcification is highly suggestive.
Diagnosis of pericardial constriction may be difficult since imaging is frequently inconclusive; pericardial calcification is highly suggestive.
In addition to Kussmaul’s sign a pericardial knock (accentuated third heart sound) may be heard. Right heart catheterization is usually required to demonstrate equalization of pressures in all four cardiac chambers and the pulmonary artery. Despite the high left-sided pressures pulmonary hypertension does not occur.
VALVULAR CARDIAC LESIONS
With the striking diminution in acute rheumatic fever the spectrum of valvular lesions has changed. MS is much less common as are combinations of lesions that used to affect several valves.
 The so-called “functional murmur,” which is not reflective of cardiac valvular disease, is a flow murmur usually originating over the pulmonic valve and heard best to the left of the sternum in the second intercostal space.
The so-called “functional murmur,” which is not reflective of cardiac valvular disease, is a flow murmur usually originating over the pulmonic valve and heard best to the left of the sternum in the second intercostal space.
The murmur is accentuated by situations in which the cardiac output is increased such as fever, pregnancy, and thyrotoxicosis.
Aortic Stenosis
 Lone AS in the sixth decade of life is most commonly associated with congenital bicuspid valves which calcify early; in the seventh decade and above atherosclerotic calcific AS in a tricuspid aortic valve is the most common cause of AS.
Lone AS in the sixth decade of life is most commonly associated with congenital bicuspid valves which calcify early; in the seventh decade and above atherosclerotic calcific AS in a tricuspid aortic valve is the most common cause of AS.
The well-known characteristic murmur of AS is harsh, located at the upper right sternal border, crescendo-decrescendo, and transmitted up the carotids and the subclavian vessels. The murmur may be present in the absence of significant obstruction (calcific aortic sclerosis). The challenge is determining, from the physical examination, whether there is significant stenosis that is associated with a substantial, hemodynamically significant, gradient across the valve.
 The most useful physical findings that suggest hemodynamically significant AS are a quiet to absent aortic closure sound (S2), a pulse delay with a slow upstroke at the carotid artery, and a late peaking murmur.
The most useful physical findings that suggest hemodynamically significant AS are a quiet to absent aortic closure sound (S2), a pulse delay with a slow upstroke at the carotid artery, and a late peaking murmur.
The left ventricle tolerates the pressure load from significant AS for substantial periods of time. Once symptoms occur the course is rapidly downhill.
 The classic symptom triad of AS is well known: angina, syncope, and CHF. The appearance of any of these heralds the need for subsequent evaluation and likely valve replacement.
The classic symptom triad of AS is well known: angina, syncope, and CHF. The appearance of any of these heralds the need for subsequent evaluation and likely valve replacement.
 There is no medical treatment for AS.
There is no medical treatment for AS.
AS is the one situation in which the cardiac output increases immediately after open heart surgery.
 Syncope in AS results from exercise-induced vasodilation in the setting of a fixed cardiac output due to the stenotic valve. CHF manifests as exertional dyspnea due to diastolic dysfunction.
Syncope in AS results from exercise-induced vasodilation in the setting of a fixed cardiac output due to the stenotic valve. CHF manifests as exertional dyspnea due to diastolic dysfunction.
 Although elderly patients with AS frequently have coexisting coronary artery disease, angina may occur with AS in the absence of coronary disease.
Although elderly patients with AS frequently have coexisting coronary artery disease, angina may occur with AS in the absence of coronary disease.
The thickened myocardium and the high intraventricular pressures in AS compromise subendocardial blood flow so that oxygen delivery cannot meet the demand imposed by exertion.
Aortic Regurgitation
 Chronic AR may be due to valvular disease or, more commonly, disease of the aortic root with dilatation and stretching of the aortic annulus.
Chronic AR may be due to valvular disease or, more commonly, disease of the aortic root with dilatation and stretching of the aortic annulus.
 Aortic root dilatation occurs with aortic aneurysms, dissections, severe hypertension, or cystic medial necrosis of the aorta.
Aortic root dilatation occurs with aortic aneurysms, dissections, severe hypertension, or cystic medial necrosis of the aorta.
The latter is the lesion of Marfan’s syndrome, but also occurs without the other Marfan’s stigmata.
 The ascultatory findings are different in valvular AR as compared with AR secondary to root dilatation. In valvular AR the aortic closure sound is usually muted and the regurgitant decrescendo murmur loudest at the left sternal border; in root disease the aortic closure sound is accentuated and may be booming while the murmur is loudest at the upper right sternal border.
The ascultatory findings are different in valvular AR as compared with AR secondary to root dilatation. In valvular AR the aortic closure sound is usually muted and the regurgitant decrescendo murmur loudest at the left sternal border; in root disease the aortic closure sound is accentuated and may be booming while the murmur is loudest at the upper right sternal border.
The “tambour” sign of aortic valve closure was classically described in syphilitic aortic aneurysm.
 The peripheral signs of AR reflect the large pulse pressure and depend upon the size of the regurgitant jet.
The peripheral signs of AR reflect the large pulse pressure and depend upon the size of the regurgitant jet.
 The water hammer (Corrigan’s or Watson’s) pulse with a rapid upswing and collapsing downswing is the easiest sign to elicit.
The water hammer (Corrigan’s or Watson’s) pulse with a rapid upswing and collapsing downswing is the easiest sign to elicit.
The water hammer pulse is recognized by holding the patient’s forearm upright and squeezing; when a clear pulse is felt the test is positive.
 In AR the precordium is active, reflecting the increased ventricular volume, while in AS the precordium is quiet.
In AR the precordium is active, reflecting the increased ventricular volume, while in AS the precordium is quiet.
In both AR and AS, left ventricular hypertrophy (LVH) is indicated by an abnormally sustained PMI; eventually, lateral displacement of the PMI occurs.
 Although chronic AR, which develops slowly, may be tolerated for significant periods of time by the gradual increase in ventricular compliance, acute AR is a medical emergency that requires immediate intervention.
Although chronic AR, which develops slowly, may be tolerated for significant periods of time by the gradual increase in ventricular compliance, acute AR is a medical emergency that requires immediate intervention.
 The major causes of acute AR are valve rupture from acute bacterial endocarditis and dissecting aneurysm of aorta.
The major causes of acute AR are valve rupture from acute bacterial endocarditis and dissecting aneurysm of aorta.
Trauma to the chest is another cause.
Mitral Stenosis
 MS and MR, in times past, were common manifestations of rheumatic heart disease. The valves themselves, inflamed by rheumatic fever, became fused in either a stenotic or open position, or commonly both.
MS and MR, in times past, were common manifestations of rheumatic heart disease. The valves themselves, inflamed by rheumatic fever, became fused in either a stenotic or open position, or commonly both.
New cases of MS are now very uncommon.
 A tipoff to the diagnosis of MS is a booming first heart sound, much easier to recognize than the classic diastolic rumble.
A tipoff to the diagnosis of MS is a booming first heart sound, much easier to recognize than the classic diastolic rumble.
 The symptoms of MS reflect either low cardiac output or pulmonary congestion.
The symptoms of MS reflect either low cardiac output or pulmonary congestion.
As left atrial pressure and pulmonary venous pressure rise pulmonary congestion and/or pulmonary hypertension develop. Depending on the degree of pulmonary hypertension, which diminishes pulmonary blood flow, either recurrent bouts of pulmonary edema or low cardiac output with right heart failure dominate the clinical picture.
 MS is classically associated with atrial fibrillation and pulmonary hypertension.
MS is classically associated with atrial fibrillation and pulmonary hypertension.
One of the observations in the early days of mitral valvulotomy, contrary to expectation, was that the pulmonary hypertension usually reversed but the atrial fibrillation did not.
Mitral Regurgitation
MR is quite common and has a diverse set of causes.
 The major current causes of MR are ischemic heart disease with papillary muscle infarction and rupture; left ventricular dilatation with distortion of the spatial relationship of the papillary muscles to each other so that contraction does not result in valve closure; mitral valve prolapse (MVP); and, in the elderly, calcification of the mitral annulus.
The major current causes of MR are ischemic heart disease with papillary muscle infarction and rupture; left ventricular dilatation with distortion of the spatial relationship of the papillary muscles to each other so that contraction does not result in valve closure; mitral valve prolapse (MVP); and, in the elderly, calcification of the mitral annulus.
 The murmur of MR is pansystolic, obliterates S2, and radiates to the axilla.
The murmur of MR is pansystolic, obliterates S2, and radiates to the axilla.
 MR remains a significant risk for the development of subacute bacterial endocarditis.
MR remains a significant risk for the development of subacute bacterial endocarditis.
 Chronic MR is better tolerated than chronic AR because there is less cardiac dilatation.
Chronic MR is better tolerated than chronic AR because there is less cardiac dilatation.
In chronic MR the low-pressure runoff into the compliant left atrium allows the left ventricular volume to diminish during systole; in chronic AR the ventricle expands during diastole because of the volume load created by the regurgitant jet from the high-pressure aorta, and cannot shrink much during systole because it is pumping into the high-pressure circuit. As a consequence the volume increases in the left ventricle are much greater with AR as compared with MR, and the adverse effects of cardiac dilatation outlined above come into play.
 Afterload reduction is an effective treatment for MR since it increases the fraction of the ventricular ejection that flows forward.
Afterload reduction is an effective treatment for MR since it increases the fraction of the ventricular ejection that flows forward.
 Acute MR, in distinction to the chronic situation, results in immediate severe pulmonary edema.
Acute MR, in distinction to the chronic situation, results in immediate severe pulmonary edema.
In acute MR, as occurs with papillary muscle rupture, the relatively noncompliant left atrium does not accommodate the regurgitant jet well, so that left atrial pressures rise dramatically and pulmonary edema ensues.
 Rupture of a chordae tendinae produces a high-pitched whiff of MR; since the regurgitant jet is much smaller, chordae tendinae ruptures are much better tolerated than papillary muscle ruptures.
Rupture of a chordae tendinae produces a high-pitched whiff of MR; since the regurgitant jet is much smaller, chordae tendinae ruptures are much better tolerated than papillary muscle ruptures.
Both papillary muscle rupture and ruptured chordae may complicate MI.
 MVP is diagnosed by maneuvers that rapidly decrease venous return, such as standing the patient up while listening over the left lower precordium; a midsystolic click and/or a murmur of MR suggest the diagnosis which can be confirmed by echocardiography.
MVP is diagnosed by maneuvers that rapidly decrease venous return, such as standing the patient up while listening over the left lower precordium; a midsystolic click and/or a murmur of MR suggest the diagnosis which can be confirmed by echocardiography.
MVP, also known as the “billowing mitral valve” or Barlow’s syndrome, is caused by myxomatous degeneration of the mitral valve leaflets.
 MVP is most common in thin, tall, young women in whom it is almost always benign unless MR is prominent and sustained. In older men it is more serious and may require valve repair.
MVP is most common in thin, tall, young women in whom it is almost always benign unless MR is prominent and sustained. In older men it is more serious and may require valve repair.
 The cause of MVP is unknown in most instances although it is sometimes associated with pectus excavatum and scoliosis. It also occurs in connective tissue diseases such as the Marfan or Ehler–Danlos syndrome.
The cause of MVP is unknown in most instances although it is sometimes associated with pectus excavatum and scoliosis. It also occurs in connective tissue diseases such as the Marfan or Ehler–Danlos syndrome.
If MR is present antibiotic prophylaxis for dental procedures is recommended.
Tricuspid Regurgitation
 Tricuspid regurgitation is almost always a consequence of pulmonary hypertension which dilates the right ventricle and stretches the annulus. Since the usual cause is left heart failure the treatment is aimed at relief of CHF; diuresis is usually effective and surgery is rarely required.
Tricuspid regurgitation is almost always a consequence of pulmonary hypertension which dilates the right ventricle and stretches the annulus. Since the usual cause is left heart failure the treatment is aimed at relief of CHF; diuresis is usually effective and surgery is rarely required.
The carcinoid syndrome and various congenital cardiac lesions may also be associated with tricuspid regurgitation.
LEFT VENTRICULAR HYPERTROPHY (LVH) AND HYPERTROPHIC CARDIOMYOPATHY (HCM)
The usual cause of LVH is increased afterload from hypertension or AS. In HCM LVH occurs without an increase in afterload.
 In LVH the PMI is sustained and displaced laterally. A fourth heart sound and left atrial enlargement on EKG are early signs of LVH.
In LVH the PMI is sustained and displaced laterally. A fourth heart sound and left atrial enlargement on EKG are early signs of LVH.
HCM is a genetic abnormality in a sarcomeric protein that causes hypertrophy and disarray of cardiomyocyte architecture.
 Asymmetric septal hypertrophy may occur as part of HCM. Contraction of the septum may cause outflow tract obstruction, also referred to as subaortic stenosis.
Asymmetric septal hypertrophy may occur as part of HCM. Contraction of the septum may cause outflow tract obstruction, also referred to as subaortic stenosis.
It is possible to distinguish outflow tract obstruction from valvular AS on PE. The aortic closure sound is not muffled and the pulse is bifid rather than plateau in outflow tract as compared with valvular obstruction; the murmur in outflow tract obstruction is loudest at the lower left sternal border and not the classic aortic area at the upper right sternal border.
 MR is frequently associated with septal hypertrophy.
MR is frequently associated with septal hypertrophy.
The characteristic murmur can be heard in the axilla.
 HCM is the leading cause of sudden death in young men.
HCM is the leading cause of sudden death in young men.
Affecting athletes in particular, ventricular cardiac arrhythmias are responsible.
 In the presence of asymmetric septal hypertrophy large septal Q waves on the EKG may be mistaken for evidence of MI.
In the presence of asymmetric septal hypertrophy large septal Q waves on the EKG may be mistaken for evidence of MI.
Infiltrative diseases affecting the myocardium may also cause the “pseudo infarction” pattern.
CONGENITAL HEART DISEASE IN ADULTS
 Ostium secundum, the most common form of atrial septal defect (ASD), is frequently diagnosed in young adults. The left to right shunt, if of sufficient size, causes diastolic overload of the right ventricle.
Ostium secundum, the most common form of atrial septal defect (ASD), is frequently diagnosed in young adults. The left to right shunt, if of sufficient size, causes diastolic overload of the right ventricle.
Exertional dyspnea, fatigue, and increased susceptibility to pulmonary infection are common manifestations.
 The physical findings are classic and include a fixed split of S2, a split M1, a systolic ejection murmur over the pulmonic valve at the upper left sternal border, and a parasternal lift indicative of right ventricular enlargement.
The physical findings are classic and include a fixed split of S2, a split M1, a systolic ejection murmur over the pulmonic valve at the upper left sternal border, and a parasternal lift indicative of right ventricular enlargement.
The left ventricle is small and the PMI is absent.
 The much less common ostium primum defect, also known as an endocardial cushion defect, is located lower down on the septum; MR is frequently associated.
The much less common ostium primum defect, also known as an endocardial cushion defect, is located lower down on the septum; MR is frequently associated.
 Chest x-ray in patients with ASD shows right ventricular hypertrophy (RVH), pulmonary vascular congestion and plethoric lungs. EKG shows a right bundle branch block (partial or complete). With the primum defect left anterior hemiblock (left axis deviation) is present as well.
Chest x-ray in patients with ASD shows right ventricular hypertrophy (RVH), pulmonary vascular congestion and plethoric lungs. EKG shows a right bundle branch block (partial or complete). With the primum defect left anterior hemiblock (left axis deviation) is present as well.
In the fetal circulation the resistance in the pulmonary circulation is very high thus shunting blood from the venous return through the foramen ovale to the left atrium and thus into the systemic circulation for oxygen exchange in the placenta. In the presence of an atrial septal defect the increased pulmonary resistance of the fetal state regresses so that pulmonary artery pressure remains relatively normal into adulthood in most cases. Eventually, the hyperdynamic pulmonary circulation causes pulmonary hypertension which, over time, becomes fixed. In the case of ventricular septal defect (VSD), if sufficiently large, the pulmonary vascular resistance never regresses and pulmonary hypertension is noted earlier in life. In many VSD cases, however, the pulmonary pressure remains normal until the hyperdynamic pulmonary circulation takes its toll (as with ASD).
 VSD is associated with a coarse, loud, pansystolic murmur at the left sternal border and a palpable thrill is present in almost all cases.
VSD is associated with a coarse, loud, pansystolic murmur at the left sternal border and a palpable thrill is present in almost all cases.
In distinction to ASD the left ventricular impulse is prominent in patients with VSD unless the defect is very small. A third heart sound may be present due to rapid left ventricular filling.
 Eisenmenger’s syndrome refers to congenital heart lesions (ASD, VSD, and patent ductus arteriosus) with reversal of the left to right shunt as pulmonary artery (PA) pressures approach systemic levels; this is associated with the development of cyanosis, clubbing, dyspnea, and occasionally, hemoptysis.
Eisenmenger’s syndrome refers to congenital heart lesions (ASD, VSD, and patent ductus arteriosus) with reversal of the left to right shunt as pulmonary artery (PA) pressures approach systemic levels; this is associated with the development of cyanosis, clubbing, dyspnea, and occasionally, hemoptysis.
Described originally with VSD it now includes, prominently, ASD with shunt reversal.
 A small VSD (“maladie de Roger”) may serve as a nidus for the development of subacute bacterial endocarditis.
A small VSD (“maladie de Roger”) may serve as a nidus for the development of subacute bacterial endocarditis.
Appropriate prophylaxis for dental and other procedures should be considered.
ORTHOSTATIC HYPOTENSION
 Orthostatic hypotension is usually defined as an upright fall in systolic BP of 20 mm Hg and a 10 mm Hg fall in diastolic pressure, with symptoms.
Orthostatic hypotension is usually defined as an upright fall in systolic BP of 20 mm Hg and a 10 mm Hg fall in diastolic pressure, with symptoms.
 Orthostatic hypotension is caused by either a decrease in plasma volume, with its attendant fall in venous return, or by a disruption of the SNS reflexes that normally defend the circulation in the upright position.
Orthostatic hypotension is caused by either a decrease in plasma volume, with its attendant fall in venous return, or by a disruption of the SNS reflexes that normally defend the circulation in the upright position.
Under normal circumstances, standing results in a fall in central venous pressure which initiates a reflex increase in sympathetic nervous system (SNS) outflow to the great veins causing venoconstriction and a corresponding increase in venous return as blood is shifted from the central venous reservoir to the heart. Cardiac output is thereby rapidly restored eliminating a significant fall in BP. Pulse rate may increase as well. Note that this reflex involves the low pressure, capacitance (venous) portion of the circulation; the classic baroreceptors in the high pressure, resistance (arterial) portion of the circulation do not come into play unless there is a fall in arterial BP. The system is designed to prevent an upright fall in BP.
 Volume depletion is the most common cause of orthostatic hypotension.
Volume depletion is the most common cause of orthostatic hypotension.
Hemorrhage, vomiting, and diarrhea are the common culprits, along with pharmaceutical agents (diuretics, antihypertensive drugs, and psychotropic agents that antagonize the SNS).
 Neurologic diseases or drugs that affect the SNS outflow or the peripheral SNS are also common causes of orthostatic hypertension.
Neurologic diseases or drugs that affect the SNS outflow or the peripheral SNS are also common causes of orthostatic hypertension.
 Multiple systems atrophy (Shy–Drager syndrome) is a neurodegenerative disease that affects multiple areas of the central nervous system; Parkinsonian features and orthostatic hypotension are prominent manifestations, the latter a consequence of involvement of the preganglionic sympathetic nerve fibers.
Multiple systems atrophy (Shy–Drager syndrome) is a neurodegenerative disease that affects multiple areas of the central nervous system; Parkinsonian features and orthostatic hypotension are prominent manifestations, the latter a consequence of involvement of the preganglionic sympathetic nerve fibers.
The disease is most common in middle-aged men.
 Degeneration of the peripheral, postganglionic, sympathetic nerves is more common in elderly women, and has orthostatic hypotension as the major symptom without the central nervous system manifestations of Shy–Drager syndrome.
Degeneration of the peripheral, postganglionic, sympathetic nerves is more common in elderly women, and has orthostatic hypotension as the major symptom without the central nervous system manifestations of Shy–Drager syndrome.
Since there is no feasible way of restoring the normal relationship between fall in venous return (and fall in BP) and sympathetic activation, treatment is generally ineffective, but measures to expand the plasma volume (high salt diet, fludrocortisone) and to antagonize venous pooling (fitted stockings or pantyhose) offer some relief in mild cases by making the circulation less dependent on SNS reflexes.
 In unexplained shock or circulatory collapse, echocardiogram of the right ventricle rapidly distinguishes between hemorrhage and pulmonary embolism; the RV is collapsed in the former and enlarged in the latter.
In unexplained shock or circulatory collapse, echocardiogram of the right ventricle rapidly distinguishes between hemorrhage and pulmonary embolism; the RV is collapsed in the former and enlarged in the latter.
SYNCOPE
Vasovagal (Neurogenic) Syncope
 Syncope, a transient loss of consciousness, is a common occurrence that usually reflects a sudden decrease in cerebral perfusion. The most common cause is a “vasovagal” reaction, also known as neurovascular or neurocardiogenic syncope.
Syncope, a transient loss of consciousness, is a common occurrence that usually reflects a sudden decrease in cerebral perfusion. The most common cause is a “vasovagal” reaction, also known as neurovascular or neurocardiogenic syncope.
The cause of vasovagal syncope is an abrupt fall in venous return which triggers a paradoxical decrease in SNS tone and an increase in vagal tone with consequent fall in BP, drop in pulse rate, and fall in cardiac output. Certain activities may trigger this response in a reproducible fashion.
 The clinical characteristics of a vasovagal faint include a warning aura of lightheadedness, ringing in the ears, nausea, and a slow fall to the ground.
The clinical characteristics of a vasovagal faint include a warning aura of lightheadedness, ringing in the ears, nausea, and a slow fall to the ground.
Once the supine position is reached venous return increases and the patient recovers consciousness.
 A primitive reflex, evolved in the setting of major trauma with hemorrhage, underlies the vasovagal reaction.
A primitive reflex, evolved in the setting of major trauma with hemorrhage, underlies the vasovagal reaction.
The underlying pathophysiology explains the triggers, the aura, and the clinical features. The usual response to a fall in venous return, as described above, is an increase in SNS activity; when the fall in venous return is abrupt and extreme, however, a primitive reflex comes into play and SNS activity markedly decreases, frequently with an associated increase in vagal tone. In teleologic terms this response would serve several useful functions in the face of severe injury such as major trauma or hemorrhage. The fall in blood pressure would limit bleeding; the decrease in metabolic rate associated with SNS suppression would conserve resources for recovery.
 The situations in which vasovagal syncope occurs are those associated with a rapid decrease in venous return such as hot day, extreme exertion, alcohol excess, or a big meal.
The situations in which vasovagal syncope occurs are those associated with a rapid decrease in venous return such as hot day, extreme exertion, alcohol excess, or a big meal.
Interestingly, some people faint at the sight of their own blood as in medical phlebotomy; this may be a CNS (cortical) representation of severe injury that elicits the same primitive response that evolved in the setting of severe trauma with bleeding.
 The fall in cerebral perfusion causes the ringing in the ears and dizziness that herald the faint.
The fall in cerebral perfusion causes the ringing in the ears and dizziness that herald the faint.
The fall in BP is gradual enough that the patient usually slumps down to the supine position without serious injury.
 If the cerebral ischemia is more profound or prolonged, seizures may ensue (convulsive syncope). These are usually in the form of myoclonic jerks that are short lived.
If the cerebral ischemia is more profound or prolonged, seizures may ensue (convulsive syncope). These are usually in the form of myoclonic jerks that are short lived.
Seizures may, of course, also be the cause of syncope.
 An elevated prolactin level frequently accompanies seizures and may be useful in reconstructing the episode if blood is available from the time of the attack. Elevation of prolactin level may also occur after some vasovagal episodes.
An elevated prolactin level frequently accompanies seizures and may be useful in reconstructing the episode if blood is available from the time of the attack. Elevation of prolactin level may also occur after some vasovagal episodes.
Although vasovagal syncope is sometimes called neurocardiogenic syncope the heart is not the culprit in this type of fainting and cardiac pacing is of no avail.
Cardiac Syncope
 Cardiac syncope is the consequence of a sudden fall in cardiac output due to valvular heart disease (particularly AS), or due to serious disturbance of cardiac rhythm.
Cardiac syncope is the consequence of a sudden fall in cardiac output due to valvular heart disease (particularly AS), or due to serious disturbance of cardiac rhythm.
Either tachy or brady arrhythmias may be associated with syncope. If the rate is rapid incomplete ventricular filling (shortened diastole) is the cause; in slow rhythms (heart block) rate-related fall in cardiac output is responsible. In AS the outflow across the aortic valve is fixed so that a fall in peripheral resistance with exercise or a decrease in cardiac filling with an increase in heart rate results in a profound fall in cardiac output and a decrease in cerebral perfusion.
In cardiac syncope loss of consciousness is sudden and damage from the fall is much more likely than with the vasovagal reaction. Aura is usually absent.
 Head injury from a sudden fall without warning is frequently the clue that the syncope is of cardiac origin.
Head injury from a sudden fall without warning is frequently the clue that the syncope is of cardiac origin.
 EKG harbingers of complete heart block are important to recognize in patients with syncope since they indicate the need for prompt placement of a cardiac pacemaker.
EKG harbingers of complete heart block are important to recognize in patients with syncope since they indicate the need for prompt placement of a cardiac pacemaker.
EKG characteristics of impending heart block include wide bundle branch block (longer than 0.12, especially if longer than 0.16), bifascicular block (such as right bundle branch block with left anterior hemiblock), and evidence of bilateral bundle branch block (left or right bundle with prolonged PR interval) and alternating bundle branch block showing a left bundle pattern in the standard leads and a right bundle pattern in the precordial leads.
 A patient presenting with a history of syncope, a bruise about the head, and EKG features suggesting bilateral bundle branch block is a candidate for immediate cardiac pacing.
A patient presenting with a history of syncope, a bruise about the head, and EKG features suggesting bilateral bundle branch block is a candidate for immediate cardiac pacing.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


