Chapter 4 The drug discovery process
General principles and some case histories
Introduction
The creation of a new drug can be broadly divided into three main phases (Figure 4.1):
• Drug discovery – from therapeutic concept to molecule
• Drug development – from molecule to registered product
• Commercialization – from product to therapeutic application to sales.
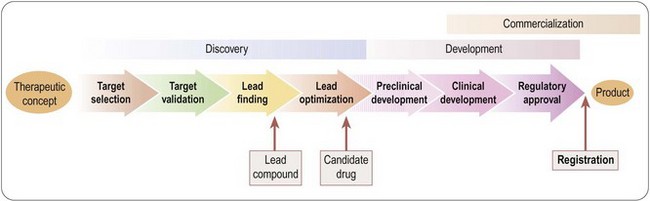
Fig. 4.1 Three main phases of the creation of a new drug: discovery, development and commercialization.
Traditionally, these functions are performed by Research, Development and Marketing, respectively, reflecting the different professional training and expertise required to do the job. Figure 4.1 greatly oversimplifies what is actually a very complex process. For example, development activities, in the form of additional clinical trials, or testing of new formulations, continue well beyond the point of registration, with the aim of extending the range of applications of the compound or complying with regulatory requirements. The discovery team, having delivered the first candidate drug, will carry on looking for others, to serve as back-ups in case the lead compound should fail in development, or as follow-up compounds intended to have advantages over the lead compound. The three components of the overall process are not independent and consecutive stages, but have to be closely coordinated at all stages of the project. At the outset of any new project, the criteria against which the plan will be judged include not only its scientific strength and originality but, importantly, development and marketing issues. For example, if the therapeutic target is an ill-defined clinical disorder, such as chronic fatigue syndrome, will it be possible to measure clinical efficacy objectively? Does the project face stiff competition from other companies working in the same area, or from drugs already in clinical use? Is it likely that an esoteric drug delivery system will be required, and if so, can this be developed? If the drug is successfully developed, is the expected market sufficient to justify the cost of development? The answers to questions of this kind are likely to change, for better or for worse, during the course of the project, so it is essential to keep such issues constantly under review, and to adapt the project plan if necessary.
To integrate successfully the different interests – and cultures – of research, development and marketing is one of the major challenges for a pharmaceutical company, and the need for such integration is a relatively modern development in the industry. As recently as 25–30 years ago in most companies, the process was much more compartmentalized: scientists produced molecules with interesting pharmacological properties, development functions were responsible for checking their safety and turning them into registrable drugs, and the marketing department generated sales and turned them into revenues. At the time this worked well, and many companies prospered. The drop-out rate was not excessive, because regulatory requirements were less stringent, and the failures that did occur were not unduly expensive in terms of time and resources lost. Since then, biomedical science has advanced dramatically, drug discovery and development have become more technology-driven and, hence, expensive, regulatory requirements are much more stringent, and the competition is more intense. With bigger teams, and more complex multidisciplinary tasks, effective project management has become much more important than it used to be to keep costs and delays to a minimum. An additional complication is that of the increased amount of work being done in partnership with other companies or with academic groups, adding the need for alliance as well as project management (see also Chapter 22).
A more detailed overview of the drug discovery phase of a typical project aimed at producing a new synthetic drug is shown in Figure 4.2. It starts with the choice of a disease area and defining the therapeutic need that is to be met. It proceeds to the identification of the biochemical, cellular or pathophysiological mechanism that will be targeted, and, if possible, the identification and validation of a molecular ‘drug target’. Next comes the identification of a lead structure, followed by the design, testing and fine-tuning of the drug molecule to the point where it is deemed suitable for development, discussed in more detail in subsequent chapters.
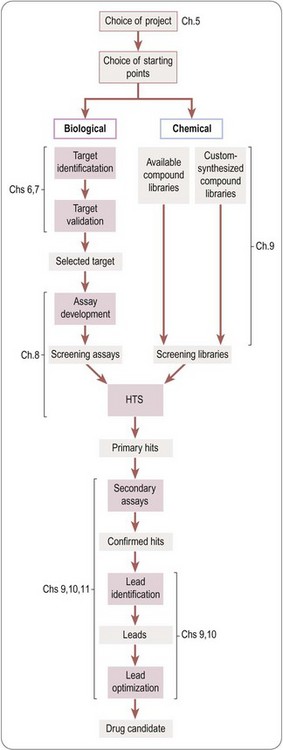
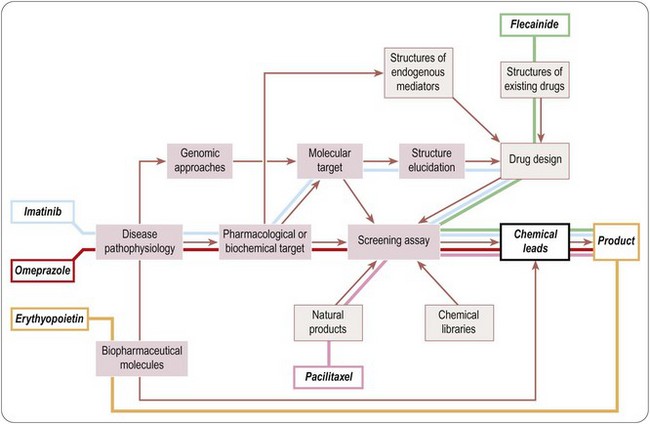
The foregoing remarks apply to the discovery of conventional ‘small-molecule’ therapeutics, and the strategy for developing biopharmaceuticals – an increasing proportion of new drugs appearing on the market – is generally different. Biopharmaceutical agents (see Chapter 3) are very diverse, including endogenous mediators, monoclonal antibodies and vaccines, and in the future, no doubt, products for siRNA and gene therapy applications. Where endogenous molecules are involved, the concept of targets and lead compounds has much less relevance, as nature has done the discovery part of the work, so once the therapeutic relevance of the substance has been established, the problems mainly revolve around the production, purification and formulation of the material in a form suitable for the market. With other kinds of biopharmaceuticals, such as therapeutic antibodies, the molecular target will generally be chosen in advance, and the main task is to obtain an antibody with the required properties.
A glance at the pharmacopoeia will show that many therapeutic agents, particularly anti-infective and anti-tumour drugs, originate from natural products, rather than synthetic molecules (Table 4.1). Until about 1950, when synthetic chemistry really came into its own as a source of new drugs, most of the pharmacopoeia consisted of natural products, and they continue to be important, as the example of paclitaxel described below shows. It is reasonable to suppose that such ready-made, highly evolved biomolecules stand a better chance of interacting with selected drug targets than do random synthetic molecules, and the pool from which they come is huge and largely untapped. Exploiting such a ready-made compound library is seen as an attractive strategy which has led to some important therapeutic breakthroughs, such as the anti-malarial drug artemesinin, immunosuppressants such as ciclosporin fujimycin (FK506) and rapamycin, as well as paclitaxel and other recently introduced anticancer drugs such as epothilones. In 2008–2010, 8 out of 64 novel compounds registered were natural products or derived from natural products. In practice, the theoretical advantages of natural products are balanced by several practical disadvantages. Access to source material in remote places can be troublesome for geographical reasons, as well as being politically sensitive, and the continuing availability of the active compound, if it cannot be synthesized on a commercial basis, may be uncertain. Microorganisms have an advantage over higher species in this regard, but initial positive test data on microbial samples frequently cannot be replicated, presumably because of inconsistencies in the culture conditions. Purification and structure determination of natural products is now fairly routine, but is often difficult and time-consuming. A recent example is the introduction of Sativex, a standardized preparation of cannabinoids, given as intra oral drops for the treatment of spasticity associated with multiple sclerosis.
Table 4.1 Examples of therapeutic drugs derived from natural products
| Warfarin | Anticoagulant. Synthetic compound derived from dicoumarol, found in spoiled sweet clover |
| Heparin | Anticoagulant, occurring naturally in mammalian tissues |
| Hirudin | Anticoagulant from leech, now produced by genetic engineering |
| Opiates | Analgesic compounds from poppies |
| Methylxanthines (caffeine, theophylline) | Phosphodiesterase inhibitors and adenosine receptor antagonists. Produced by tea, coffee and coca plants |
| Statins | HMG CoA reductase inhibitors used to reduce plasma cholesterol. Lovastatin is a fungal metabolite. Later compounds (mevastatin, pravastatin) synthesized from lovastatin |
| Cromoglycate | Asthma prohylaxis. Synthetic compound based on khellin, a plant product used as a herbal medicine |
| Vinca alkaloids (vincristine, vinblastine) | Anticancer drugs produced by plants of the periwinkle family |
| Paclitaxel | Anticancer drug from yew tree |
| Etoposide | Anticancer drug synthesized from podophyllotoxin, produced by mandrake plant; used in folk medicine |
| Artemether | Antimalarial drug, semisynthetic derivative of artemesin, produced by Chinese herb |
| Ivermectin | Antihelminthic drug, semisynthetic derivative of avermectin, a fungal metabolite |
| Antibiotics | Too numerous to list. The majority of current antibiotics are derived from fungal metabolites |
In earlier times the pharmacopoeia consisted very largely of plant-derived compounds (e.g. opiates, atropine, ephedrine, ergot alkaloids, strychnine, tubocurarine, digoxin, quinine, veratridine, reserpine, etc.), many of which remain in therapeutic use or provide valuable research tools.
Some case histories
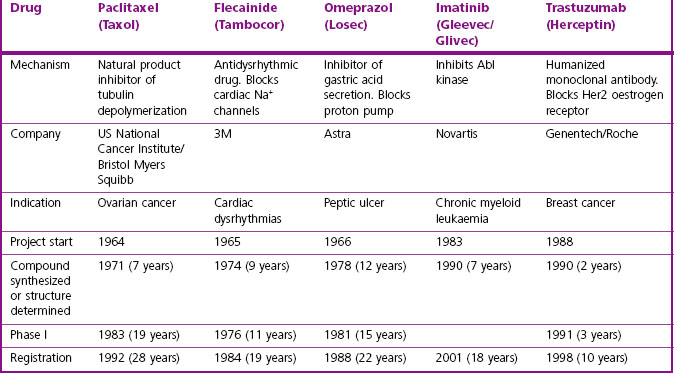
It is clear that there are many starting points and routes to success in drug discovery projects (Lednicer, 1993; Drews, 2000). The brief case histories of five successful drugs, paclitaxel (Taxol), flecainide (Tambocor), omeprazol (Losec), imatinib (Gleevec/Glivec) and trastuzumab (Herceptin), are summarized in Figure 4.3 and Table 4.2, and described in more detail below. Each represents a highly innovative ‘breakthrough’ project rather than an incremental development based on an existing therapy, and they illustrate the variety of different approaches taken by successful projects over the past 35 years. However, for several reasons we should avoid interpreting these as guidelines for success in the future. For one thing, the approach changes as the underlying technologies advance; furthermore, pharmaceutical companies generally publicize only their successes, and even then the accounts are often somewhat sanitized, and fail to describe the errors that were made, the deadlines missed and the blind alleys that were encountered – the full ‘shaggy drug stories’ generally remain discreetly hidden. It must be remembered that, of drug discovery projects begun, only about 1 in 50 is successful in terms of bringing a compound to market. Only at the point when official approval for trials in man is granted does the project become visible to the outside world, so data on success rates, timelines, etc., are much more accessible for the minority of projects that progress to Phase I or beyond than for the majority that never get that far. Analysing the success factors for early-stage drug discovery projects is therefore difficult.
Paclitaxel (Taxol)
Paclitaxel is an interesting example of a project based on the development of a natural product (Cragg, 1998). It began in the early 1960s, when the US National Cancer Institute, responding to the Nixon-inspired ‘war on cancer’, set up one of the first directed screening programmes – still running – to seek new anticancer drugs from plant sources. The sample of bark from the Pacific Yew was collected in 1962 and found to have modest activity against various tumour cell lines. The active substance was isolated in 1969 and joined a collection of moderately active, but not particularly interesting, lead compounds. When this collection was dusted off in 1975 and tested on a new assay, a melanoma cell line, paclitaxel stood out as highly active. Its activity was confirmed in animal models, and it was soon chosen as a development candidate. Interest was further stimulated when its novel mechanism of action, the promotion of microtubule polymerization, was very elegantly demonstrated. Development was difficult, for two main reasons. Paclitaxel is insoluble in water, and the early formulations for injection used in Phase I trials contained a high proportion of the solubilizing agent Cremophor EL, causing frequent severe allergic reactions when given as a bolus intravenous injection. After considerable delay, the problem was overcome by the use of slow infusions and development was resumed. The second problem was the supply of material for clinical trials, and the uncertainty that it could ever be produced on a commercial basis. The Pacific Yew grows slowly and has a restricted habitat, and conservationists were opposed to commercial harvesting. As a result, there was only enough material for limited Phase II studies, on patients with ovarian cancer. The improvement in these patients was dramatic, but continuation of the project, now in collaboration with Bristol Myers Squibb, was seriously hindered by the limited supplies of yew tree bark. The conservation concerns were overcome when a census showed that the tree population was not in fact threatened, and industrial supplies of bark were collected to support the trials programme right through to 1992, when the drug was officially approved.
Flecainide (Tambocor)
The story of flecainide (Banitt and Schmid, 1993) represents a completely different route to success, variations of which gave rise to many innovative drugs (e.g. antihypertensive drugs, antidepressants and antipsychotics) during the 1960s. In the early 1960s, the drugs used to treat cardiac dysrhythmias were mainly quinidine, procainamide, digoxin (for supraventricular tachycardias) and lidocaine (given i.v. for ventricular dysrhythmias). The first three had many troublesome side effects, whereas lidocaine’s use was largely confined to intensive care settings. In 1964, the 3M company decided to seek better antidysrhythmic drugs. Their chemists had developed a new synthetic pathway for introducing –CF3 groups, and they started a chemistry programme based on fluorinated derivatives of known local anaesthetic and antidysrythmic drugs. Assays for antidysrhythmic activity at the time involved elaborate studies on anaesthetized dogs, which were quite unsuitable for screening, and so the group developed a simple primary screening assay based on the ability of compounds to prevent ventricular fibrillation induced by chloroform inhalation in mice, which was used to screen hundreds of compounds. Secondary assays on selected compounds were carried out on anaesthetized dogs in the then conventional fashion. Questions of mechanism were not addressed, it being (correctly) assumed that efficacy in these animal models would serve as a good predictor of clinical efficacy irrespective of the cellular mechanisms involved. A potential development compound was synthesized in 1969, but abandoned on account of CNS side effects. After a further 5 years of painstaking chemistry, during which many different structural classes were tested, flecainide was synthesized (1974) and found to have a much improved therapeutic window compared to its predecessors. The first clinical studies were performed in 1976, and development proceeded quite smoothly until the compound was registered in 1984. It was the first deliberate effort to develop an improved antidysrhythmic drug and proved highly successful in the clinic, now accepted as the standard Class 1c antidysrhythmic agent according to the current classification.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


