CHAPTER 28 The chronic lymphoid leukemias
In this chapter we deal with the chronic lymphoproliferative disorders that usually present with lymphocytosis (i.e. as leukemia). We discuss these conditions using the newest edition of the World Health Organization (WHO 2008) classification as a framework;1 bone marrow (BM) involvement by lymphoid neoplasms more often presenting as lymphoma is described in Chapter 29.
Chronic lymphocytic leukemia
B-cell chronic lymphocytic leukemia (CLL) is a chronic leukemia resulting from the proliferation of a neoplastic clone of monoclonal B-lymphocytes with a very characteristic immunophenotype (CD19+/CD5+/CD23+).The CLL diagnosis can be readily established if these cells are more than or equal to 5 × 109/l in peripheral blood (PB).1
Clinical features
CLL is the most common leukemia in the Western world with an incidence of the order of 6/100 000/year, whereas the disease is much less prevalent in Asian countries.2 It is mainly a disease of the middle-aged and elderly, and with a male : female ratio of approximately 2 : 1 (http://www.hmrn.org/Statistics/Incidence.aspx).
Pathologic features
Peripheral blood
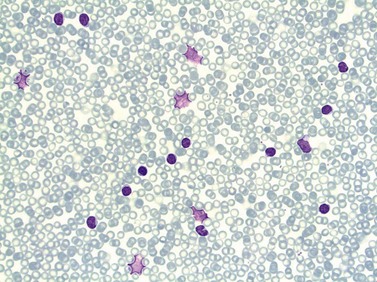
In early-stage disease, the only PB abnormality is lymphocytosis with an increase of mature small lymphocytes, which are relatively uniform in their cytologic features (Fig. 28.1). The lymphocytes typically have a high nucleocytoplasmic ratio, condensed chromatin and an inapparent or barely apparent nucleolus. Sometimes the chromatin is condensed into a mosaic pattern. Smear cells are typically seen in blood films, but are not pathognomonic (Fig. 28.1). The presence of some plasmacytoid lymphocytes and small numbers of cells with cleft or irregular nuclei is compatible with a diagnosis of CLL. There may be up to 10% prolymphocytes (atypical cells with larger, more prominent nucleoli). The presence of more than 10% prolymphocytes or of a spectrum of cells from small to large, with cytoplasmic basophilia, may be associated with increased proliferation and disease progression. If the number of prolymphocytes exceeds 55% a diagnosis of prolymphocytic leukemia should be considered.
Bone marrow
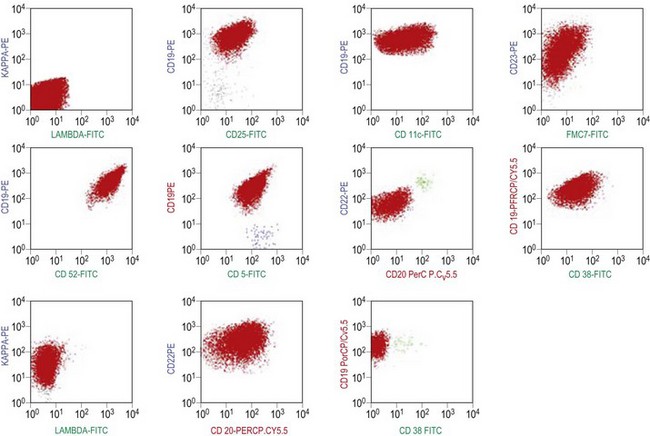
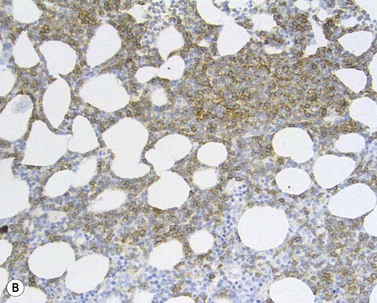
The BM aspirate is hypercellular as the result of an increase in small mature lymphocytes with the same cytologic features as those in blood. A threshold of 30% lymphocytes has been previously applied as a diagnostic criterion of CLL involvement in BM aspirate; nowadays it is usually replaced by detection of monoclonal B-cell population >5 × 109/l with CLL phenotype by flow cytometry (FCM) (Fig. 28.2).
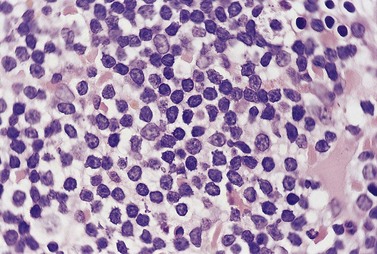
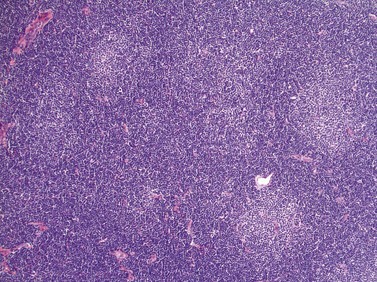
The BM trephine biopsy (BMTB) shows variable degree of infiltration. The neoplastic infiltrate is composed predominantly of small lymphocytes with low numbers of prolymphocytes and para-immunoblasts (Fig. 28.3). The small lymphocytes have coarsely clumped chromatin and scanty cytoplasm. Prolymphocytes are slightly larger than small lymphocytes with a dispersed chromatin pattern and a small central nucleolus. Para-immunoblasts are medium-sized cells with an open chromatin pattern, prominent central nucleolus and moderate amounts of basophilic cytoplasm. Prolymphocytes and para-immunoblasts may be present in greater numbers in some areas of the infiltrate. Those areas usually show higher proliferation and are called proliferation centers (Fig. 28.4A). Four patterns of infiltration are seen – interstitial, nodular, nodular-interstitial and diffuse. The interstitial pattern is characterized by neoplastic cells infiltrating individually between normal hemopoietic precursors and fat cells. Nodular infiltrates focally replace fat and hemopoietic cells. Nodular-interstitial infiltration is a combination of the nodular and interstitial patterns (Fig. 28.4B). In cases with diffuse infiltration there is complete replacement of hemopoietic precursors and fat cells. Unlike many other BM lymphoid infiltrates, there is usually little or no increase in reticulin associated with infiltration by CLL.
Other tissues
Lymph node involvement in CLL is characterized by effacement of nodal architecture by a diffuse infiltrate of small lymphocytes with the formation of proliferation centers in which there are increased numbers of prolymphocytes and para-immunoblasts. At low power proliferation centers appear paler than the surrounding areas, and if numerous can give the infiltrate an appearance of nodularity (Fig. 28.5).
In the spleen, infiltration leads to expansion of the white pulp, with some cases also having involvement of the red pulp.3 Infiltration of the liver involves both portal tracts and sinusoids. Rarely there may be symptomatic infiltration of the skin,4 CNS5 or prostate.6 In all these tissues the infiltrate is made up predominantly of small lymphocytes.
Rapid development of an extramedullary tumor is the typical presentation of Richter’s syndrome, which represents the clinico-pathologic transformation of CLL to an aggressive lymphoma, most commonly diffuse large B-cell lymphoma (DLBCL).7 In most cases, CLL transform to a clonally related DLBCL, but development of a DLBCL unrelated to the CLL clone has also been described.7 In some patients the infiltration by CLL and DLBCL can be seen in the same biopsy material. In rare patients, a transformation towards Hodgkin lymphoma has been described and Epstein–Barr virus has been implicated in pathogenesis of this transformation.8
Immunophenotype
Besides characteristic CD19/CD5/CD23 phenotype, FCM studies show a weak surface membrane expression of monotypic (kappa or lambda) immunoglobulin, usually IgM with or without IgD and a weak expression of certain B-cell markers, specifically CD20, CD22 and CD79b9 (Fig. 28.2). FMC7, which is expressed by most other mature B-cell neoplasms, is usually weak or negative, while CD43 and CD200 are positive. CD11c and CD25 are variably expressed.
Modern FCM technology can now detect low levels of cells with CLL phenotype in 0.6–12%10 of healthy population with a male : female ratio of approximately 2 : 1. The prevalence increases with age and in individuals with first-degree relatives with CLL. The absolute numbers of cells with CLL phenotype are low (median 13, range 3–1458 per mm3) and represent a minor proportion of total B-lymphocytes in most cases (5–10%).10 It has been demonstrated that virtually all cases of CLL have been preceded by CLL-type MBL several years prior to diagnosis.11 However, the risk for an individual with CLL-type MBL to develop CLL is likely to be low and currently estimated as approximately 1% per year.
Cytogenetics and molecular genetics
In recent years, molecular genetic studies have revealed new prognostic markers which have significantly improved the subdivision of CLL. One of the most stable molecular markers, the somatic hypermutation status of the immunoglobulin heavy variable (IGHV) genes, divides CLL into two clinical subgroups, where patients with unmutated IGHV genes (≥98% identity to the corresponding germline gene; ~30–40% of patients) show a considerably worse prognosis than patients with mutated IGHV genes (<98% identity to the corresponding germline gene; 60–70% of patients).12,13 IGHV unmutated CLL patients also display a more progressive disease, more frequently carry high-risk genomic aberrations and require chemotherapy at an earlier stage than IGHV mutated CLL patients. IGHV mutational analysis can be performed at any time point during the disease course, since the mutational status will remain unchanged in CLL.
Immunogenetic studies have suggested that antigen stimulation may be involved in disease development.14 This is supported by the finding of non-random combinations of specific IGHV-D-J genes and homologous complementarity determining region 3 (CDR3) leading to structural similarity of the B-cell receptor (BCR) in a significant proportion of CLL patients.15,16 The similarity of the BCR among patients belonging to a ‘stereotyped’ subset suggests that the antigens these receptors bind to are similar and potentially relevant to disease pathogenesis. Interestingly, it was recently indicated that stereotyped subsets may not only share biological but also clinical features.16 For instance, the IGHV3-21/IGLV3-21 subset has been associated with a poor outcome irrespective of IGHV gene mutational status.
Potential surrogate markers for the IGHV mutational status have been suggested, e.g. the expression levels of CD38 and ZAP70 as determined by flow cytometry.13,17 Although studies have shown an insufficient correlation between the CD38 level and IGHV mutational status, CD38 may still serve as an independent prognostic factor.18 More promising is the expression level of the tyrosine kinase ZAP70, which predict the IGHV gene mutation status with high accuracy,17,19 although discordant results have been reported.18
In contrast to many other B-cell lymphoma entities, there are no genetic aberrations that are common to all CLL patients. Several recurrent aberrations have been characterized, where the most frequent are deletion of 13q14 (50–55%), deletion of 11q22-23 (12–18%), trisomy 12 (11–16%) and deletion of 17p13 (5–10%).20 These recurrent aberrations are often present only in a proportion of cells of the leukemic clone, indicating that they do not reflect the initial leukemogenic event. The 11q22-23 deletion encompasses the ATM gene and the 17p13 deletion covers the TP53 gene, where both of these genes are crucial in maintaining cell cycle control. Interestingly, two micro-RNAs, miR-15 and miR-16, have recently been shown to be encoded in the deleted region of 13q, and have been proposed to be of pathogenetic importance in CLL.21
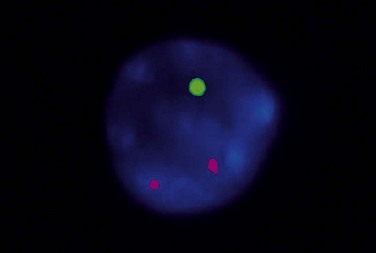
Patients with 13q deletion, if present as a single aberration, are known to have a better outcome compared to patients with 11q and especially 17p deletions, whereas trisomy 12 patients show a more intermediate risk-profile.20 Patients with poor-risk aberrations also respond poorly to current treatment protocols, in particular the 17p-deleted subgroup. Since conventional cytogenetics give a low success rate (will only detect ~40–50% aberrations), fluorescence in situ hybridization (FISH) analysis has became the gold standard in CLL using a commercially available panel of FISH probes to screen for 11q-, 13q-, +12 and 17p-. This FISH analysis is usually performed on interphase cells from a peripheral blood smear where the proportion of aberrant cells is counted (Fig. 28.6). Since clonal evolution can occur in CLL and high-risk aberrations can emerge over time, it is important to perform FISH analysis if there is a change in clinical course or if the patient does not respond to therapy as expected.
Leukemic presentations of other B-cell lymphomas
Blood and BM presentatations of other B-cell lymphomas are described in detail in Chapter 29. Frequency of leukemic presentation and immunophenotypes of cells that can be found in blood and bone marrow are summarized in Table 29.1.
Prolymphocytic leukemias
B-lineage prolymphocytic leukemia
Clinical features
B-PLL is usually a disease of the elderly (median age 70 years) with a slight male preponderance. Typically, there is splenomegaly with only minor lymphadenopathy. In most patients the rate of disease progression is much more rapid than that of CLL. A paraprotein is present more often than in CLL. Prognosis is considerably worse than that of CLL with the median survival being about 3 years.22
Pathologic features
Peripheral blood
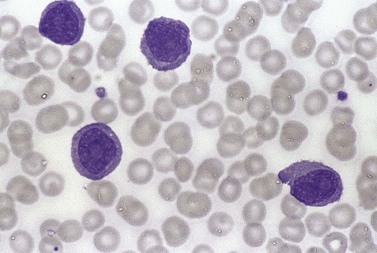
The white cell count is usually markedly elevated as the result of the presence of considerable numbers of abnormal lymphoid cells. Prolymphocytes are medium-sized to large cells, characterized by a prominent nucleolus which appears vesicular because of perinucleolar chromatin condensation (Fig. 28.7). In some patients, the majority of neoplastic cells are typical large prolymphocytes with large prominent nucleoli. In others, there is a spectrum of cells with the medium-sized and smaller cells having less prominent nucleoli. Prolymphocytes must exceed 55% of blood cells. Smear cells are not a typical feature. Anemia and thrombocytopenia are common.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


