46
Targeted Anticancer Therapies
This chapter will be most useful after having a basic understanding of the material in Chapter 62 Targeted Therapies: Tyrosine Kinase Inhibitors, Monoclonal Antibodies, and Cytokines, and Chapter 63 Natural Products in Cancer Chemotherapy: Hormones, in Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition. In addition to the material presented here, the 12th Edition contains:
• A discussion of the use of targeted anticancer therapies in combination with cytotoxic agents in Chapter 60 General Principles of Cancer Chemotherapy
• The molecular structures of small molecule drugs used in targeted anticancer therapies
LEARNING OBJECTIVES
 Understand the mechanisms of action and clinical uses of small molecule inhibitors of protein tyrosine kinases.
Understand the mechanisms of action and clinical uses of small molecule inhibitors of protein tyrosine kinases.
 Understand the mechanisms of action and clinical uses of monoclonal antibodies that target growth factor receptors and other tumor cell antigens.
Understand the mechanisms of action and clinical uses of monoclonal antibodies that target growth factor receptors and other tumor cell antigens.
 Understand the mechanisms of action and clinical uses of monoclonal antibodies that target the VEGF pathway.
Understand the mechanisms of action and clinical uses of monoclonal antibodies that target the VEGF pathway.
 Understand the mechanism of action and clinical uses of anticancer drugs that target the mTOR pathway, and proteosome-mediated protein degradation.
Understand the mechanism of action and clinical uses of anticancer drugs that target the mTOR pathway, and proteosome-mediated protein degradation.
 Understand the mechanism of action and clinical uses of anticancer drugs that target the IL-2 receptor.
Understand the mechanism of action and clinical uses of anticancer drugs that target the IL-2 receptor.
 Understand the clinical uses of the immunomodulatory analogs (IMiDs) in treating multiple myeloma and myelodysplastic syndrome.
Understand the clinical uses of the immunomodulatory analogs (IMiDs) in treating multiple myeloma and myelodysplastic syndrome.
 Understand the mechanism of action and use of hormone therapy to treat hormone-dependent tumors.
Understand the mechanism of action and use of hormone therapy to treat hormone-dependent tumors.
 Know the common and important toxicities of targeted anticancer pharmacotherapies.
Know the common and important toxicities of targeted anticancer pharmacotherapies.
 Know mechanisms of acquired resistance to specific targeted anticancer therapies.
Know mechanisms of acquired resistance to specific targeted anticancer therapies.
DRUGS INCLUDED IN THIS CHAPTER
• 131I-Tositumomab (BEXXAR)
• 90Y-Ibritumomab tiuxetan (ZEVALIN)
• Abarelix (PLENAXIS; withdrawn from market)
• Abiraterone (ZYTIGA)
• Aldesleukin (IL-2; PROLEUKIN)
• Alemtuzumab (CAMPATH)
• Anastrozole (ARIMIDEX)
• Bevacizumab (AVASTIN)
• Bicalutamide (CASODEX, others)
• Bortezomib (VELCADE)
• Buserelin (SUPREFACT)—not available in the United States
• Cetrorelix (CETROTIDE)
• Cetuximab (ERBITUX)
• Cyproterone—not available in the United States
• Dasatinib (BMS-354825; SPRYCEL)
• Degarelix (FIRMAGON)
• Denileukin diftitox (ONTAK)
• Deslorelin—not available in the United States
• Dexamethasone
• Erlotinib (TARCEVA)
• Everolimus (AFINITOR)
• Exemestane (AROMASIN)
• Flutamide (EULEXIN)
• Fulvestrant (FASLODEX)
• Ganirelix—not available in the United States
• Gefitinib (IRESSA)
• Gemtuzumab ozogamicin (MYLOTARG)—withdrawn from the US market in 2010
• GnRH (GONADORELIN)
• Goserelin (ZOLADEX)
• Histrelin (VANTAS)
• Imatinib mesylate (STI 571; GLEEVEC, GLIVEC)
• Ketoconazole (NIZORAL)
• Lapatinib (TYKERB)
• Lenalidomide (REVLIMID)
• Letrozole (FEMARA)
• Leuprolide (LUPRON, ELIGARD)
• Medroxyprogesterone (DEPO-PROVERA, others)
• Megestrol (MEGACE)
• Megestrol acetate (MEGACE, others)
• Nafarelin (SYNAREL)
• Nilotinib (AMN107; TASIGNA)
• Nilutamide (NILANDRON)
• Ofatumumab (ARZERRA)
• Panitumumab (VECTIBIX)
• Pazopanib (VOTRIENT)
• Prednisone
• Raloxifene (EVISTA)
• Ranibizumab (LUCENTIS)
• Rapamycin (sirolimus; RAPAMUNE)
• Rituximab (RITUXAN)
• Sorafenib (NEXAVAR)
• Sunitinib (SUTENT)
• Tamoxifen (NOLVADEX, others)
• Temsirolimus (TORISEL)
• Thalidomide (THALOMID)
• Toremifene (FARESTON)
• Trastuzumab (HERCEPTIN)
• Triptorelin (TRELSTAR, TRELSTARDEPOT)
MECHANISMS OF ACTION AND RESISTANCE OF TARGETED ANTICANCER DRUGS
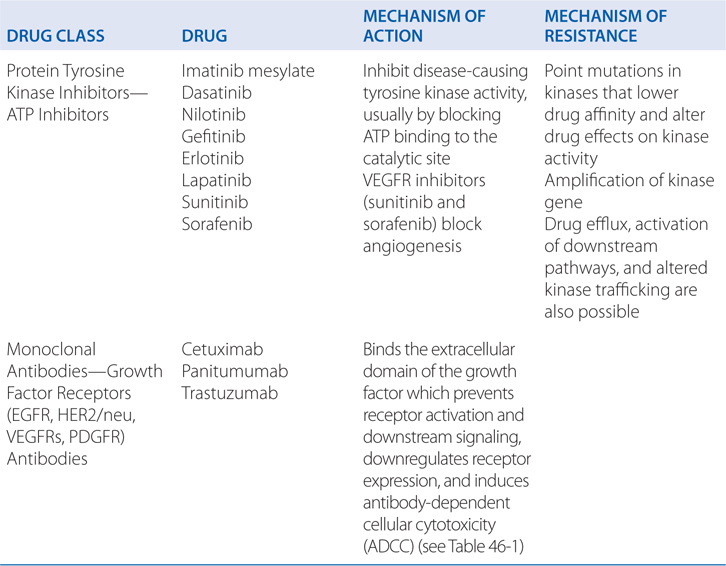
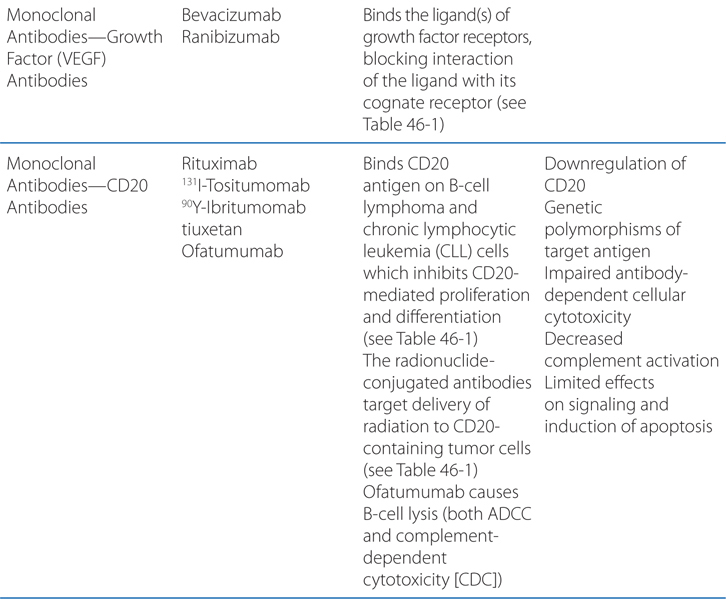
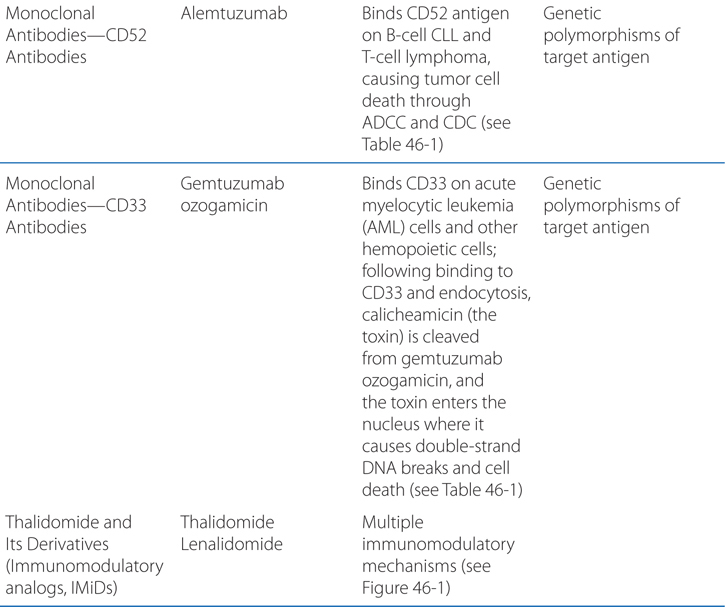

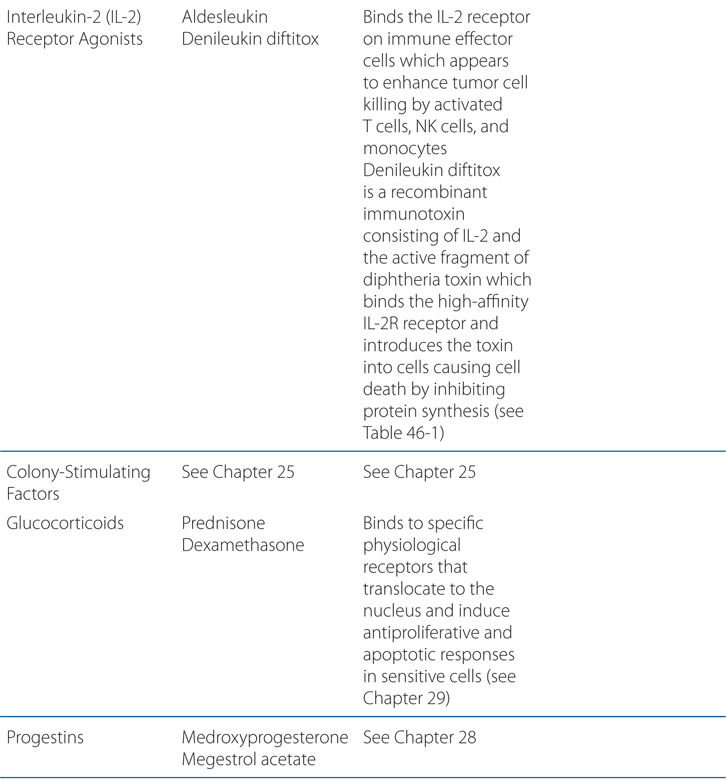
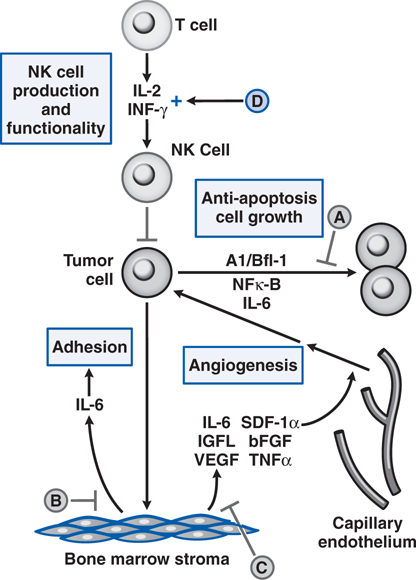
FIGURE 46-1 Schematic overview of proposed mechanisms of antimyeloma activity of thalidomide and its derivatives. Some biological hallmarks of the malignant phenotype are indicated in the boxes. The proposed sites of action for thalidomide (letters inside circles) are hypothesized to also be operative for thalidomide derivatives. A. Direct anti–multiple myeloma (MM) effect on tumor cells, including G1 growth arrest and/or apoptosis, even against MM cells resistant to conventional therapy. This is due to the disruption of the antiapoptotic effect of BCL-2 family members, blocking NF-κB signaling, and inhibition of the production of interleukin-6 (IL-6). B. Inhibition of MM-cell adhesion to bone marrow stromal cells partially due to the reduction of IL-6 release. C. Decreased angiogenesis due to the inhibition of cytokine and growth factor production and release. D. Enhanced T-cell production of cytokines, such as IL-2 and interferon-γ (IFN-γ), that increase the number and cytotoxic functionality of natural killer (NK) cells. VEGF, vascular endothelial growth factor.
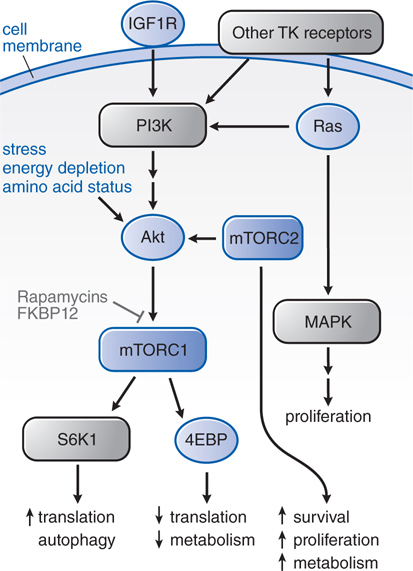
FIGURE 46-2 Insulin-like growth factor 1 receptor (IGF-1R) and other tyrosine kinase (TK) growth factor receptors signal through multiple pathways. A key pathway is regulated by phosphatidylinositol-3 kinase (PI3K) and its downstream partner, the mammalian target of rapamycin (mTOR). Rapamycins complex with FKBPP12 to inhibit the mTORC1 complex. mTORC2 remains unaffected and responds by upregulating Akt, driving signals through the inhibited mTORC1. The various downstream outputs of the 2 complexes are shown. Phosphorylation of 4EBP by mTOR inhibits the capacity of 4EBP to inhibit eif-4E and slow metabolism. 4EBP, eukaryotic initiation factor 4e (eif-4E) binding protein; FKBP12, the immunophilin target (binding protein) for tacrolimus (FK506); S6K1, S6 kinase 1.
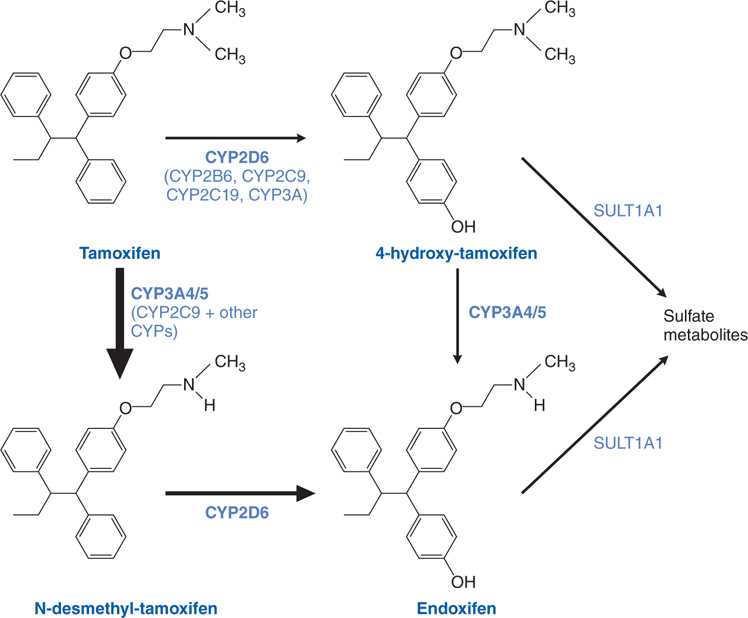
FIGURE 46-3 Tamoxifen and its metabolites.
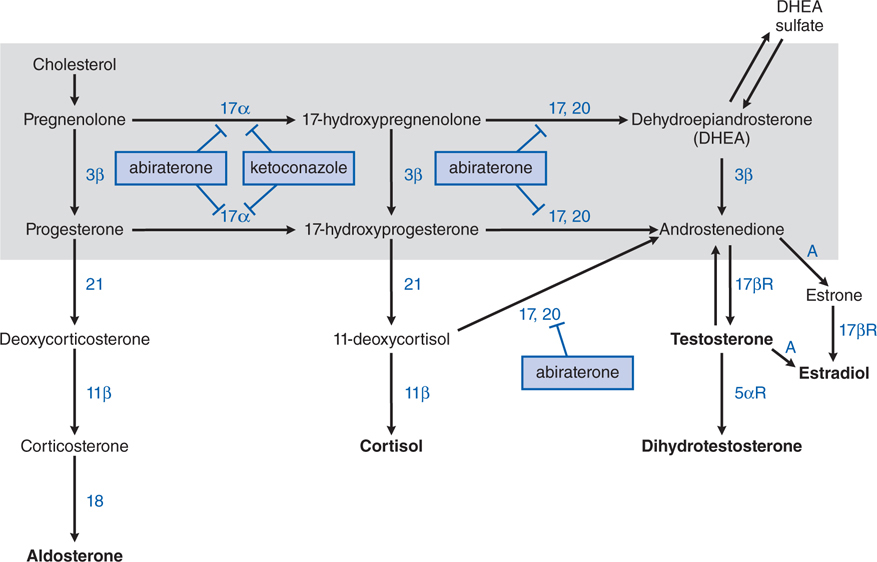
FIGURE 46-4 Steroid synthesis pathways. The enclosed shaded area contains the pathways used by the adrenal glands and gonads. Enzymes are shown next to their respective biochemical pathways, inhibitors are shown in boxes with T-shaped arrows pointing at the enzymes they inhibit. 11β, 11β-hydroxylase; 17,20, C-17,20-lyase (also CYP17); 17α, 17α-hydroxylase (CYP17); 17βR, 17β-reductase; 18, aldosterone synthase; 21, 21-hydroxylase; 3β, 3β-hydroxysteroid dehydrogenase; 5αR, 5α-reductase; A, aromatase.
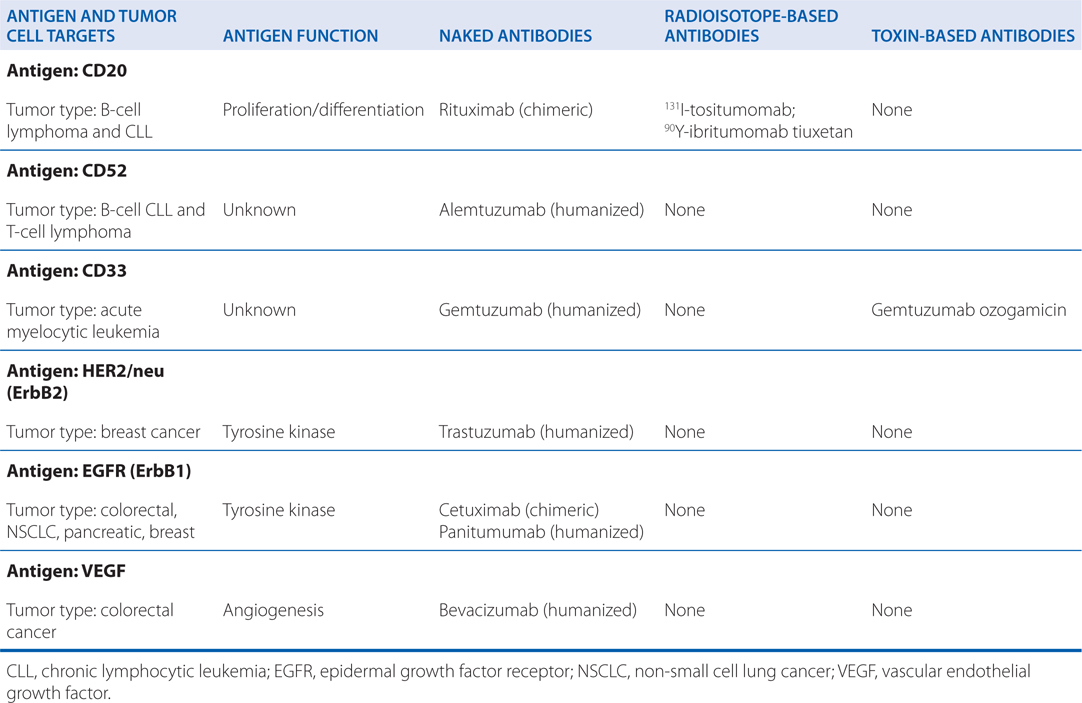
TABLE 46-1 Monoclonal Antibodies Approved for Hematopoietic and Solid Tumors
A 34-year-old man is diagnosed with Philadelphia chromosome-positive (Ph+) chronic myelogenous leukemia (CML).
a. What is the molecular mechanism that causes this form of cancer?
A single molecular event, in this case the 9:22 chromosomal translocation which results in the Philadelphia chromosome (Ph+), leads to expression of the Abelson protooncogene kinase ABL fused to BCR (breakpoint cluster region), yielding a constitutively activated protein tyrosine kinase, BCR-ABL, and then the malignant phenotype. This is the most common mechanism causing CML.
b. What is the first-line treatment for CML and what is the rationale for this pharmacotherapy?
Imatinib and the related compounds dasatinib and nilotinib induce clinical and molecular remissions in more than 90% of CML patients in the chronic phase of disease. These agents target the BCR-ABL tyrosine kinase and inhibit its activity.
c. What are the mechanisms of resistance to imatinib pharmacotherapy of CML?
Resistance to the tyrosine kinase inhibitors arises from point mutations in 3 separate segments of the BCR-ABL kinase domain. The contact points between imatinib and the enzyme become sites of mutations in drug-resistant leukemic cells; these mutations prevent tight binding of the drug and lock the enzyme in its open configuration, in which it has access to substrate. Most such mutations hold the enzyme in its open and enzymatically active confirmation. The most common resistance mutations affect amino acids 255 and 315, both of which serve as contact points for imatinib; these mutations confer high-level resistance to imatinib and nilotinib. Dasatinib is unaffected by mutation at 255 but is ineffective in the presence of mutation at 315. Nilotinib retains inhibitory activity in the presence of most point mutations (except at 315) that confer resistance to imatinib.
Other mutations affect the phosphate-binding region and the “activation loop” of the domain with varying degrees of associated resistance. Some mutations, such as at amino acids 351 and 355, do not affect response to dasatinib or nilotinib but confer low levels of resistance to imatinib.
Molecular studies of circulating tumor cells have detected resistance-mediating kinase mutations prior to initiation of therapy, particularly in patients with Ph+ acute lymphoblastic leukemia (ALL) or CML in blastic crisis. This finding strongly supports the hypothesis that drug-resistant cells arise through spontaneous mutation and expand under the selective pressure of drug exposure. Mutations may become detectable in the peripheral blood of patients receiving imatinib in the accelerated phase and in the late (>4 years from diagnosis) chronic phase of CML, heralding the onset of drug resistance.
Mechanisms other than BCR-ABL kinase mutation play a minor role in resistance to imatinib. Amplification of the wild-type kinase gene, leading to overexpression of the enzyme, has been identified in tumor samples from patients resistant to treatment. The multidrug resistant (MDR) gene, which codes for a drug efflux protein, confers resistance experimentally but has not been implicated in clinical resistance. Finally, Philadelphia chromosome-negative clones lacking the BCR-ABL translocation and displaying the karyotype of myelodysplastic cells may emerge in patients receiving imatinib for CML and may progress to myelodysplasia (MDS) and to acute myelocytic leukemia (AML). Their origin is unclear.
d. What other cancers and diseases are effectively treated with imatinib?
Imatinib has efficacy in diseases in which the ABL, kit, or PDGFR protein kinases have dominant roles in driving the proliferation of the tumor, reflecting the presence of a mutation that results in constitutive activation of the kinase, either by fusion with another protein or via point mutations. Thus, imatinib shows remarkable therapeutic benefits in patients with chronic-phase CML (BCR-ABL), GIST (kit mutation-positive gastrointestinal stromal tumor), chronic myelomonocytic leukemia (EVT6-PDGFR translocation), hypereosinophilia syndrome (FIP1L1-PDGFR), and dermatofibrosarcoma protuberans (constitutive production of the ligand for PDGFR). It is the agent of choice for GIST patients with metastatic disease and as adjuvant therapy of c-kit-positive GIST. GIST biology is particularly instructive, as patients with an exon 11 mutation of kit have a significantly higher partial response rate (72%) than those with no detectable kit mutations (9%).
e. What are the common and important side effects of therapy with imatinib, dasatinib, and nilotinib?
Imatinib, dasatinib, and nilotinib cause GI distress (diarrhea, nausea, and vomiting), but these symptoms usually are easily controlled. All 3 drugs promote fluid retention, which may lead to dependent edema, and periorbital swelling. Dasatinib may cause pleural effusions. Nilotinib may prolong the QT interval, and should be used with caution in patients with underlying heart disease or arrhythmias, although ventricular arrhythmias have not been reported. Significant myelosuppression occurs infrequently but may require transfusion support, dose reduction, or discontinuation of the drug. All 3 drugs in this class can be associated with hepatotoxicity. Most nonhematological adverse reactions are self-limited and respond to dose adjustments. After the adverse reactions such as edema, myelosuppression, or GI symptoms have been resolved, the drug may be reinitiated and titrated back to effective doses.
In a number of epithelial cancers, the epidermal growth factor receptor (EGFR) is overexpressed or is activated by mutations.
a. What role does the EGFR play in these epithelial cancers?
The EGFR belongs to the ErbB family of transmembrane receptor tyrosine kinases. EGFR, also known as ErbB1 or HER1, is essential for the growth and differentiation of epithelial cells. Ligand binding to the extracellular domain of EGFR family members causes receptor dimerization and stimulates the protein tyrosine kinase activity of the intracellular domain, resulting in autophosphorylation of several Tyr residues in the C-terminal domain. Recognition of the phosphotyrosines by other proteins initiates protein-protein interactions that result in stimulation of a variety of signaling pathways, including MAPK, PI3K/Akt, and STAT pathways (see Figures 46-2 and 46-5). In epithelial cancers, overexpression (or mutational activations) of the EGFR is a common finding and, to some extent, creates a dependence on EGFR signaling in these tumors.
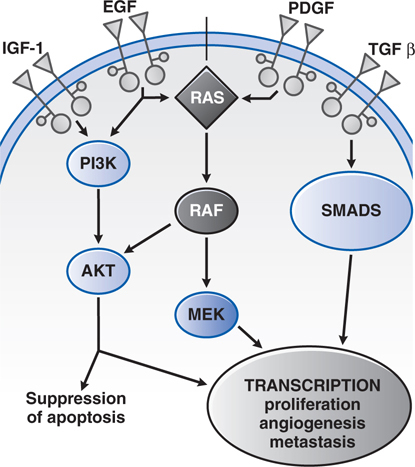
FIGURE 46-5 Growth factor signaling. Binding of agonist ligands to growth factor receptors causes receptor dimerization and activation of cytosolic protein kinase domains, leading to activation of multiple signaling pathways. Shown here are the RAS/MAPK/ERK, PI3K, and SMAD pathways, each of which is activated by receptors or cross-talk from adjacent pathways. Their signals regulate proliferation, metabolism, survival, and the synthesis of other growth factors, such as the vascular endothelial growth factor (VEGF).
b. What drugs are available to target the EGFR in epithelial cancers?
Two separate classes of drugs that target the EGFR pathway have become important agents in the therapy of solid tumors. The EGFR tyrosine kinase inhibitors erlotinib and gefitinib bind to the kinase domain and block the enzymatic function of EGFR. The monoclonal antibodies cetuximab and panitumumab (see Table 46-1) bind specifically to the extracellular domain of EGFR. They inhibit EGFR-dependent signaling through inhibition of ligand-dependent activation and receptor dimerization, downregulation of EGFR expression, and induction of antibody-dependent cell-mediated cytotoxicity.
c. What kinds of epithelial cancers are effectively treated with EGFR-targeted agents?
Erlotinib is approved for first-line treatment of patients with locally advanced, unresectable, or metastatic pancreatic cancer in combination with gemcitabine, and is also approved for second-line treatment of patients with locally advanced or metastatic non–small cell lung cancer.
Gefitinib initially was approved for the third-line treatment of patients with non–small cell lung cancer based on promising results in 2 small clinical trials. However, a larger, randomized, placebo-controlled trial failed to show an effect on survival, leading the FDA to restrict its use to patients who have previously received clinical benefit from the drug. Gefitinib continues to be widely used outside the United States.
Retrospective analysis of multiple studies has revealed that patients who were nonsmokers, Asians, or women were most likely to respond to gefitinib. Tumors from these patients frequently have characteristic activating mutations in EGFR. These mutations mostly fall into 2 groups: (1) small in-frame deletions within exon 19 and (2) the L858R point mutation. Trials that randomized never- and light-smoking patients to first-line gefitinib or to chemotherapy demonstrated a 70% response rate to gefitinib in patients with an activating EGFR mutation, compared with responses in only 1% in patients without an EGFR mutation. In patients with EGFR-mutant tumors, the response rate to gefitinib was double the response to standard chemotherapy. Therefore, mounting evidence supports the use of gefitinib, and potentially erlotinib, as first-line therapy in patients selected for the presence of sensitizing EGFR mutations.
Cetuximab won FDA approval based on an improvement in overall survival when used in combination with radiation therapy for locally or regionally advanced squamous cell carcinoma of the head and neck (HNSCC). It received a second indication as monotherapy for patients with metastatic or recurrent HNSCC who had failed platinum-based chemotherapy. It has become a useful agent in combination with cisplatin-based chemotherapy, where it has shown an improvement in survival compared to chemotherapy alone.
Panitumumab improves progression-free survival in patients with metastatic colorectal carcinoma, as demonstrated in patients with EGFR-expressing tumors who had 2 or more previous therapies.
d. What are the common and important toxicities of EGFR-targeted agents?
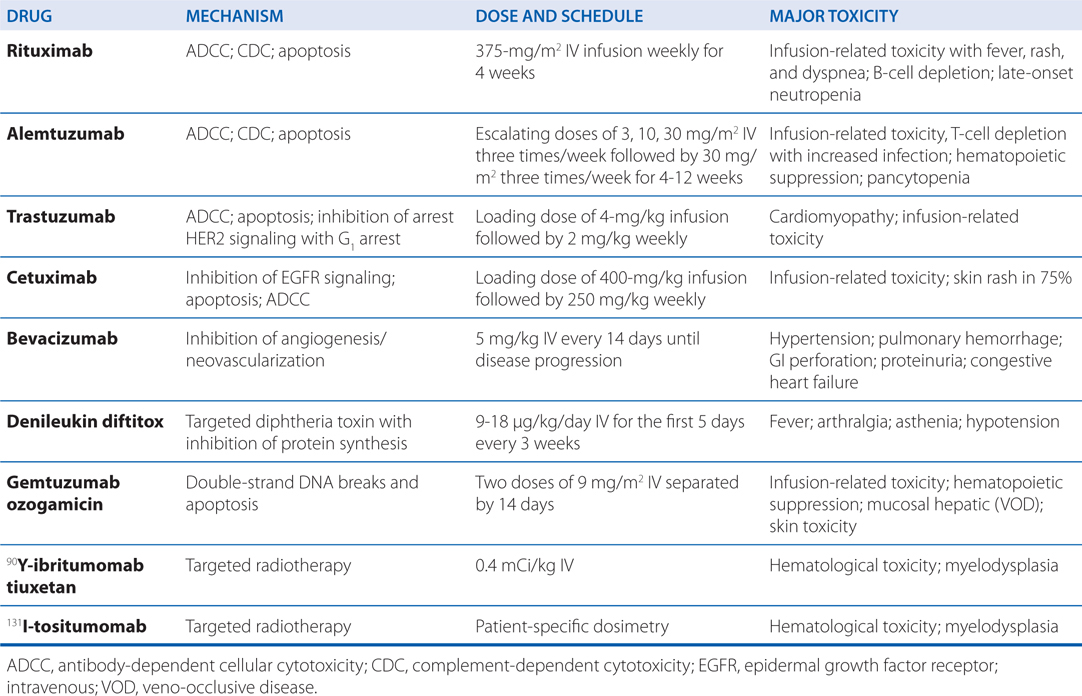
The most common adverse reactions in patients receiving erlotinib and gefitinib are diarrhea, an acneform rash, anorexia, and fatigue. Most adverse effects occur within the first month of therapy and are tolerable when managed with supportive medications and dose reductions. Cetuximab therapy is associated with infusion-related toxicity and skin rash in 75% of treated patients (see Table 46-2).
TABLE 46-2 Dose and Toxicity of Monoclonal Antibody-Based Drugs
Serious or fatal interstitial lung disease, often associated with symptoms of cough and dyspnea, occurs with a frequency of 0.7 to 2.5%.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


