Chapter 3 SYSTEMIC DISEASES
Introduction

 Pathogenesis—how did it develop.
Pathogenesis—how did it develop.
 Pathology—what anatomic changes it produced.
Pathology—what anatomic changes it produced.
 Pathophysiology—what functional consequences it has.
Pathophysiology—what functional consequences it has.
 Clinical features—what signs and symptoms it produced.
Clinical features—what signs and symptoms it produced.
 Treatment and outcome—how to cure it and how to predict the final prognosis.
Treatment and outcome—how to cure it and how to predict the final prognosis.
Systemic diseases are classified according to their pathogenesis as follows:







In this chapter we discuss several diseases that could be used as prototypes for all other diseases in each of these eight categories. We use hereditary hemochromatosis as an example of a genetic disease because it illustrates how a mutation of a gene encoding a single protein (HFE—involved in regulating the intestinal absorption of iron) can affect multiple organ systems. We discuss gout as an example of a metabolic disease and show how the same metabolic abnormality (hyperuricemia) has several causes and still produces the same set of clinical signs and symptoms. As an example of toxic diseases we discuss alcohol abuse. Alcoholism demonstrates how substance abuse can cause major health problems. The complexities of circulatory disturbances are discussed in the example of shock and closely related multiple organ failure. Acquired immunodeficiency syndrome (AIDS) is our example of a systemic infectious disease, illustrating the consequences of the primary viral infection and bacterial, viral, fungal, and parasitic superinfections that complicate this disease complex. As an example of autoimmune disorders we examine systemic lupus erythematosus (SLE), an immune complex-mediated hypersensitivity reaction that can affect numerous organs. Finally, the systemic effects of a malignant tumor are illustrated by discussing carcinoma of the lung, not only because it is the number one cause of cancer-related death, but also because of its multiple systemic effects.
Acquired immunodeficiency syndrome (AIDS) Immunodeficiency induced by human immunodeficiency virus (HIV) infection, characterized by a low count of helper T cells (CD4+ cells) and opportunistic infections involving the skin, FI system, lungs, or the central nervous system.
Alcoholism Pattern of alcohol intake resulting in psychophysical dependence on alcohol and dysfunction in one or more of five aspects of living: marital, social, legal, occupational, or physical.
Autoimmune diseases Group of chronic diseases characterized by abnormal immune reactions to self antigens. Such diseases may be accompanied by overproduction of autoantibodies or abnormal T-cell reactions, and pathological or functional changes in various organs.
Carcinoma Malignant neoplasm of epithelial origin.
Gout Metabolic disease characterized by hyperuricemia and a deposition of monosodium urate crystals in joints and soft tissues.
Hereditary hemochromatosis Common genetic disease associated with abnormal intestinal absorption of iron and iron overload that damages several vital organs, most notably the liver, islets of the pancreas and other endocrine organs, the heart, and the skin.
Hypersensitivity Increased reactivity to exogenous or endogenous antigens, characterized by an overproduction of antibodies or abnormal cell-mediated reactions. Hypersensitivity to foreign antigens is also called allergy.
Hyperuricemia Increased concentration of uric acid in blood in excess of the empirically determined upper limit of normal (>7 mg/dL for females, >8 mg/dL for males); it may be asymptomatic or it may be lead to a deposition of urate crystals in tissues, resulting in gout, urate nephropathy, or urolithiasis.
Sepsis Presence of infectious pathogens or their derivatives in blood. Clinical consequences of sepsis are called septicemia and include fever, loss of vascular tone, and at least some signs of shock and multiple organ failure.
Shock Set of systemic circulatory and metabolic disturbances involving numerous body functions caused by inadequate blood perfusion of vital organs.
Systemic lupus erythematosus (SLE) Multisystem autoimmune disease of unknown origin characterized by the formation of circulating immune complexes and their deposition in numerous anatomic sites.
Hereditary Hemochromatosis
Unregulated absorption of iron in the duodenum leads to iron overload.
Under normal circumstances iron is absorbed in the duodenum mostly in form of ferrous (Fe2+) iron (Fig. 3-1). The absorption of iron depends on the iron stored in the body, but normally enterocytes in the duodenum absorb only 10% of the total amount of iron entering the duodenum (i.e., 1–2 mg, from the 10–20 mg of iron ingested) daily in the typical American diet.
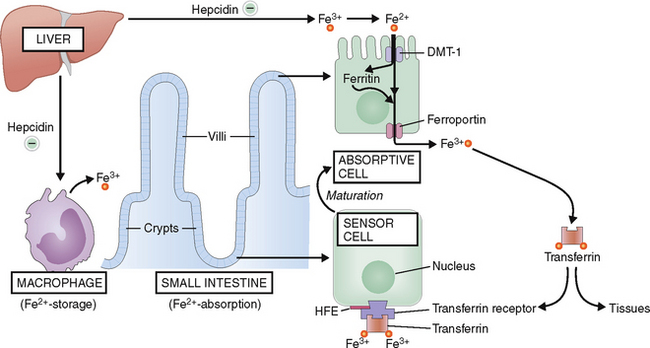
In hereditary hemochromatosis the mutation of HFE leads to an uncontrolled absorption of iron in the duodenum. The total iron stores, which are normally around 2.5 g for women and 3.5 g for men can be increased 10 to 20 times and even more. The saturation of the iron transport protein transferrin is also increased, and the excess iron is also excreted into the urine.



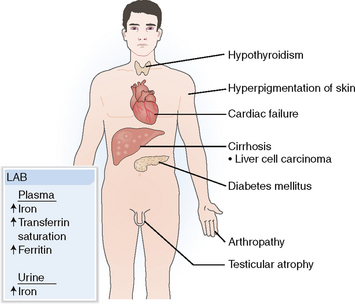
Iron overload causes pathologic changes in several vital organs.
Hemochromatosis may damage many organs, but most often the pathologic changes are seen in the liver (95%), skin (90%), pancreas (65%), joints (35%), and the heart (15%) (Fig. 3-2). Clinical symptoms usually manifest after age 40. Symptoms are 5 to 10 times less common in women, because normally women lose blood during menstruation and are thus less prone to accumulate iron. The symptoms occur later and are typically encountered several years after the onset of menopause.
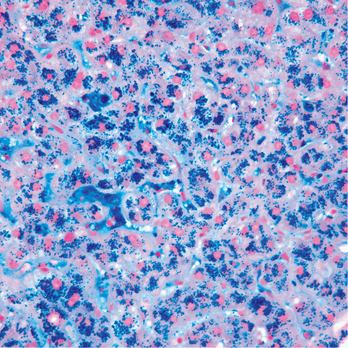
Liver. Iron accumulates in Kupffer cells, hepatocytes, and even in bile duct cells (Fig. 3-3). Deposits of iron pigment lead to fibrosis, gradually progressing to frank cirrhosis. Clinically these changes are associated with hepatomegaly, portal hypertension with splenomegaly, and esophageal varices. Other signs and symptoms of liver failure are also present in advanced cases. Liver cell carcinoma develops in 20% to 30% of patients with cirrhosis.
Endocrine glands. Accumulation of hemosiderin in the pancreas is associated with diabetes and related to injury of the islets of Langerhans. Diabetes mellitus develops more readily in persons who have a genetic predisposition and a family history of diabetes. Other endocrine organs may be affected as well, most notably the thyroid and the gonads. Testicular atrophy and consequences of reduced testosterone production (e.g., loss of libido, erectile dysfunction, gynecomastia) are common.
Hemochromatosis is associated with diagnostic changes in iron metabolism.
 Total plasma iron. Normal plasma contains iron in a concentration of 50 to 170 μg/dL (9–30 μm/L). In hereditary hemochromatosis plasma iron concentration is usually increased to over 200 μg/dL.
Total plasma iron. Normal plasma contains iron in a concentration of 50 to 170 μg/dL (9–30 μm/L). In hereditary hemochromatosis plasma iron concentration is usually increased to over 200 μg/dL.


 Urinary iron excretion. Normally the urine collected over 24 hours contains less than 2 mg of iron. In hereditary hemochromatosis urinary excretion is increased at least five times over the normal limit.
Urinary iron excretion. Normally the urine collected over 24 hours contains less than 2 mg of iron. In hereditary hemochromatosis urinary excretion is increased at least five times over the normal limit.
> Serum transferrin saturation over 50% in a fasting person is highly suggestive of hemochromatosis.
Deposits of iron in the liver can be estimated subjectively in liver biopsy specimens stained with Prussian blue reacting with hemosiderin (see Fig. 3-3). Furthermore, the iron content of the liver biopsy specimen can be quantitated biochemically and expressed as iron in milligrams per gram of liver tissue. Liver containing increased amounts of iron appears denser than normal on CT or MRI examination. Genetic testing provides the final diagnosis. Genetic testing should also be offered to first-degree relatives. Treatment includes weekly phlebotomy to reduce the total iron content of the tissues.
Gout
 Only 2% to 3% of adults develop gout, even though 10% of the total adult population has hyperuricemia, defined empirically as an elevation of uric acid concentration in blood over 7 mg/dL (0.41 mmol/L). There is a sex-related difference in uric acid metabolism, and thus the actual upper limit of normal is 7 mg/dL for females and 8 mg/dL for males.
Only 2% to 3% of adults develop gout, even though 10% of the total adult population has hyperuricemia, defined empirically as an elevation of uric acid concentration in blood over 7 mg/dL (0.41 mmol/L). There is a sex-related difference in uric acid metabolism, and thus the actual upper limit of normal is 7 mg/dL for females and 8 mg/dL for males.


Uric acid concentration in blood depends on the balance between production and excretion of uric acid.
Uric acid is the end product of the degradation of adenine and guanine. The immediate metabolic precursor of uric acid is xanthine (Fig. 3-4). Xanthine may be produced directly from guanine or from hypoxanthine through the action of xanthine oxidoreductase. This enzyme has a dual function, acting as oxidase and dehydrogenase. It also transforms xanthine into uric acid. Although the breakdown of nucleic acids occurs in all tissues, uric acid is produced only in organs, such as the liver and the intestines, that contain xanthine oxidoreductase.
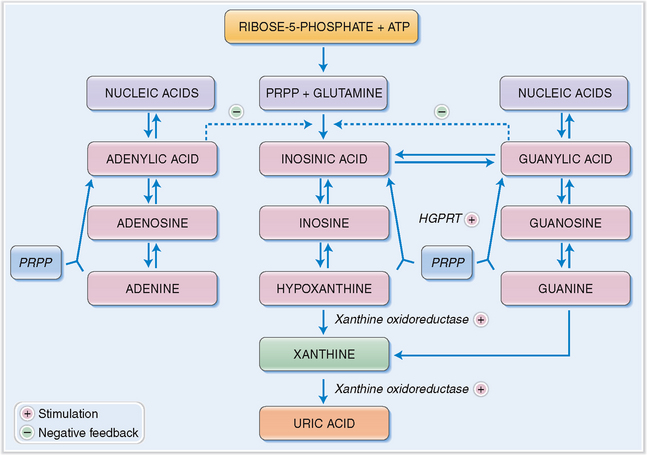
The body contains approximately 1800 mg of uric acid, one third of which is turned over daily. Approximately two thirds of all uric acid is derived from the degradation of the purines, adenine and guanine (Fig. 3-5). The remaining third is derived from the diet. Two thirds of uric acid is eliminated from the blood in urine, and the remaining third through the intestines.
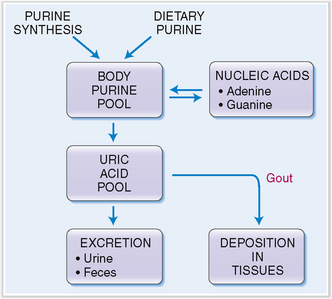
Ionized uric acid binds to sodium, and in blood it is predominantly found in the form of monosodium urate. Under physiologic conditions monosodium urate is saturated at a concentration of 6.8 mg/dL (415 μmol/L), and at higher concentrations one would expect it to precipitate and form crystals. This, however, does not occur, because the blood contains some stabilizing substances that prevent such crystallization. The solubility of monosodium urate decreases at low temperature, which accounts for the deposition of urate crystals in the joint tissue of the big toe during attacks of gouty arthritis.
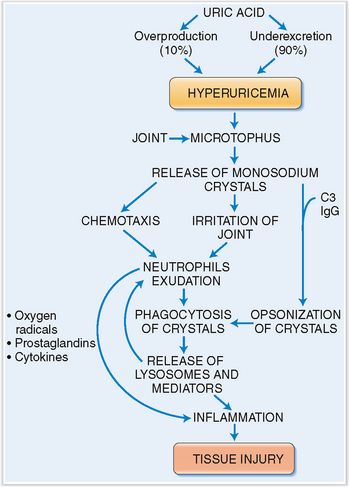
Hyperuricemia may develop due to overproduction or underexcretion of uric acid.
 Primary uric acid overproduction. The prototype of this condition is Lesch-Nyhan syndrome. This inborn error of purine metabolism related to the deficiency of HGPRT is a rare X-linked cause of primary hyperuricemia. The deficiency of HGPRT results in a dysfunction of the salvage pathway and an overproduction of uric acid and gout of early onset. Several other genetic diseases also produce hyperuricemia, but fortunately these conditions are rare.
Primary uric acid overproduction. The prototype of this condition is Lesch-Nyhan syndrome. This inborn error of purine metabolism related to the deficiency of HGPRT is a rare X-linked cause of primary hyperuricemia. The deficiency of HGPRT results in a dysfunction of the salvage pathway and an overproduction of uric acid and gout of early onset. Several other genetic diseases also produce hyperuricemia, but fortunately these conditions are rare.
 Secondary hyperproduction of uric acid. Uric acid overproduction typically occurs in tumor lysis syndrome after chemotherapy. Chemotherapy-induced killing of tumor cells results in a release of purine and pyrimidines from damaged nuclei; purines are then metabolized into uric acid. Leukemia, lymphoma, and chronic hemolytic anemia also cause hyperuricemia. Chronic diseases characterized by epithelial proliferation—such as psoriasis or excessive bone formation, such as occurs in Paget’s disease of bones—are also associated with overproduction of uric acid.
Secondary hyperproduction of uric acid. Uric acid overproduction typically occurs in tumor lysis syndrome after chemotherapy. Chemotherapy-induced killing of tumor cells results in a release of purine and pyrimidines from damaged nuclei; purines are then metabolized into uric acid. Leukemia, lymphoma, and chronic hemolytic anemia also cause hyperuricemia. Chronic diseases characterized by epithelial proliferation—such as psoriasis or excessive bone formation, such as occurs in Paget’s disease of bones—are also associated with overproduction of uric acid.




Deposits of uric acid crystals in the joints cause inflammation.
Patients who have hyperuricemia tend to form deposits of monosodium phosphate in the connective tissue capsule of joints. These aggregates, called microtophi, may then spontaneously release monosodium urate into the synovial fluid, or a larger bolus of it is released due to mechanical trauma. Crystals in the synovial fluid have an irritating effect on the synovium and also have a chemotactic effect on neutrophils, which enter the joint cavity, thus contributing to the inflammation. The crystals also become coated with complement and immunoglobulin from exudated plasma. Complement and immunoglobulin act as opsonins, thus enabling the neutrophils to phagocytize the crystals. Activated neutrophils also produce oxygen radicals, arachidonic acid derivatives, and cytokines, which stimulate inflammation, cause vasodilation, and increase vascular permeability. These events account for the redness of the joint and its swelling. Crystals ingested into the phagocytic vacuoles of neutrophils tend to pierce the lysosomes, thus allowing a discharge of lytic enzyme into the extracellular space. Once initiated, the inflammation is autocatalytic and self-sustained, lasting for several days until it slowly abates (Fig. 3-6).
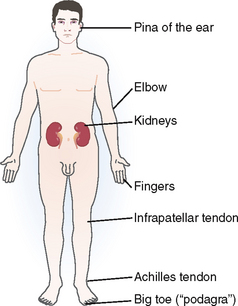
Chronic deposits of uric acid crystals lead to the formation of tophi.
Microscopic aggregates of monosodium urate crystals tend to enlarge with time until they transform into nodular masses visible by the naked eye or in radiographs in the periarticular bone. These nodules are called tophi (from the Latin word tophus, meaning porous stone). Most often they are found around the joints and the subchondral bone or subcutaneous tissue (Fig. 3-7). Typical locations are the metatarsophalangeal joint of the big toe, Achilles and infrapatellar tendons, the elbow and fingers, or the pinna of the ear. Tophi may form also in internal organs, such as the kidneys, but less commonly than in subcutaneous sites and around joints.
Hyperuricemia may damage kidneys and cause nephrolithiasis.
Prolonged hyperuricemia may affect the function of renal tubules (urate nephropathy). Deposits of monosodium phosphates are found in the medulla, where they may cause tissue injury and predispose a person to secondary infection (pyelonephritis). Uric acid stones are formed in acid urine. The incidence of calcium phosphate stones is also increased.
Alcoholism
The following definitions are useful for considering the adverse effects of alcohol:
Alcohol dependence. This term is used to describe uncontrollable alcohol intake associated with tolerance to the effects of alcohol and symptomatic withdrawal when alcohol is not available. The diagnosis is made if at least three of the criteria listed in Table 3-1 are met.
Table 3-1 Criteria for the Diagnosis of Alcohol Dependence
Modified from Greene HL, Fincher RM, Johnson WP (eds): Clinical Medicine, 2nd ed. St. Louis, Mosby, p. 748, 1996.
Alcohol has direct effects on the brain.
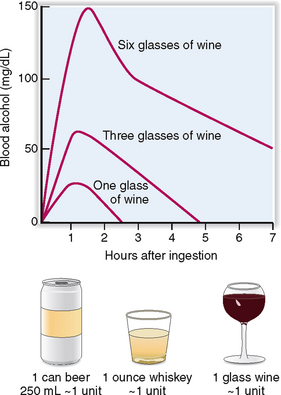
Alcohol acts on brain cells as a toxin. It changes the fluidity of cell membranes, affecting the transmission of neural impulses, thus causing depression of neural activity. Alcohol has a sedative effect on most neural centers, but the suppression of some centers may lead to a loss of inhibition of others. This may induce a feeling of relaxation or good mood, as well as increased motor activity or uncontrolled emotions. The effects are dose-dependent and can be predicted roughly from the blood alcohol concentration (Fig. 3-8) as follows:

 50 to 150 mg/dL (11–33 mmol/L)—loss of motor coordination
50 to 150 mg/dL (11–33 mmol/L)—loss of motor coordination
 150 to 200 mg/dL (33–43 mmol/L)—delirium
150 to 200 mg/dL (33–43 mmol/L)—delirium
 300 to 400 mg/dL (65–87 mmol/L)—unconsciousness and coma or respiratory arrest
300 to 400 mg/dL (65–87 mmol/L)—unconsciousness and coma or respiratory arrest
Alcohol is metabolized in the liver.
Alcohol is absorbed in the stomach and small intestine, from which it is transported into the liver. In liver cells alcohol is metabolized through the action of alcohol dehydrogenase in the cytosol, P450 cytochrome (CYP2E) in the smooth endoplasmic reticulum of the microsomal fraction, and catalase in peroxisomes (Fig. 3-9).
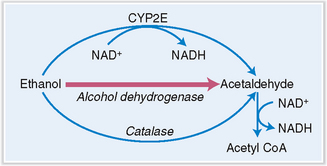
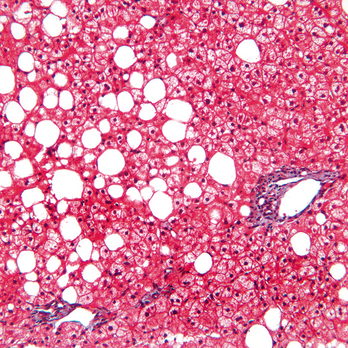
Alcohol dehydrogenase is the most important liver enzyme involved in the oxidation of alcohol. It oxidizes alcohol into acetaldehyde, a toxic metabolite, which is in turn oxidized to acetyl coenzyme A by aldehyde dehydrogenase. Nicotine adenine dinucleotide (NAD+) is the cofactor for both oxidation reactions and in this process is reduced to NADH. An increased ratio of NADH to NAD+ inhibits the NAD+-dependent oxidation of lactate to pyruvate, leading to lactic acidosis. A lack of pyruvate may cause hypoglycemia, which is further exacerbated by poor intake of nutrients in chronic alcoholics. Beta oxidation of fatty acid is reduced and triglyceride formation enhanced, leading to fatty change in liver cells (Fig. 3-10). Hence, alcohol metabolism in the liver ultimately results in the formation of toxic products and metabolic disturbances affecting the function of liver cells. If these changes persist, fatty liver may progress into alcoholic steatohepatitis and finally into cirrhosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


